06 November 2020: Review Articles
Mitochondrial Fusion and Fission in Neuronal Death Induced by Cerebral Ischemia-Reperfusion and Its Clinical Application: A Mini-Review
Yike Chen1BC*, Songxue Guo2D, Yajuan Tang1E, Chaohui Mou3F, Xinben Hu1C, Fangjie Shao1BD, Wei Yan1A, Qun Wu1AGDOI: 10.12659/MSM.928651
Med Sci Monit 2020; 26:e928651






Abstract
ABSTRACT: Mitochondria are highly dynamic organelles which are joined by mitochondrial fusion and divided by mitochondrial fission. The balance of mitochondrial fusion and fission plays a critical role in maintaining the normal function of neurons, of which the processes are both mediated by several proteins activated by external stimulation. Cerebral ischemia-reperfusion (I/R) injury can disrupt the balance of mitochondrial fusion and fission through regulating the expression and post-translation modification of fusion- and fission-related proteins, thereby destroying homeostasis of the intracellular environment and causing neuronal death. Furthermore, human intervention in fusion- and fission-related proteins can influence the function of neurons and change the outcomes of cerebral I/R injury. In recent years, researchers have found that mitochondrial dysfunction was one of the main factors involved in I/R, and mitochondria is an attractive target in I/R neuroprotection. Therefore, mitochondrial-targeted therapy of the nervous system for I/R gradually started from basic study to clinical application. In the present review, we highlight recent progress in mitochondria fusion and fission in neuronal death induced by cerebral I/R to help understanding the regulatory factors and signaling networks of aberrant mitochondrial fusion and fission contributing to neuronal death during I/R, as well as the potential neuroprotective therapeutics targeting mitochondrial dynamics, which may help clinical treatment and development of relevant dugs.
Keywords: Hypoxia-Ischemia, Brain, Nuclear Fission, Brain Ischemia, Cell Death, Mitochondrial Dynamics, Neurons
Introduction to Cerebral Ischemia-Reperfusion
The term ischemia, denoting deficient blood supply to tissues due to obstruction of the arterial inflow, was first used in the early nineteenth century. Thereafter, researchers have tried their best to discover the underlying mechanisms of ischemia-induced tissue damage for more than 200 years, hoping to developing therapies to limit the devastating health and economic burdens imposed by disorders characterized by reductions in organ-specific blood flow [1]. Cerebral ischemia-reperfusion (I/R) injury is one of the major worldwide causes of brain injury among patients with cerebrovascular diseases [2]. Timely and efficient blood flow reperfusion can rescue the endangered neurons and brain tissues and improve the prognosis of patients. With the improvement of medical technology, more effective therapeutics of stroke have emerged such as tissue plasminogen activator (tPA) and surgical intervention aiming to resupply blood flow for ischemic tissues. However, restoring blood flow following a certain ischemia period can also trigger I/R injury via the death of neurons and hence aggravate brains dysfunction. There are still no effective methods to prevent I/R injury, as the detailed underlying mechanisms of I/R injury are not completely understood. Further work on the molecular pathological process would provide new clues for developing novel effective therapy for clinical applications. Therefore, in the present review we discuss mitochondria fusion and fission in neuronal death induced by cerebral ischemia-reperfusion based on recent relevant publications and provide new insight into its potential application for clinical practice.
Mitochondrial Dynamics
Mitochondria are highly dynamic organelles undergoing coordinated cycles of fission and fusion, referred to as ‘mitochondrial dynamics’, in order to maintain their shape, distribution, and size [3]. Mitochondria are mobile organelles; their morphology is in dynamic equilibrium with the balance of fission and fusion. They are usually elongated bodies of filamentous or rod-like shape, although granular mitochondria are also encountered and vary in size from cell to cell [4]. Generally, mitochondria have 3 critical roles in cells: 1) producing ATP through glycolysis to meet with the energy demands of cells; 2) connecting to the endoplasmic reticulum to regulate calcium concentration; and 3) reactive oxygen species (ROS) management, and programmed cell death. Neurons are polymorphic cells, characterized by long axons and numerous but short dendrites. Interestingly, the mitochondria in neurons are distinct from others. Neurons need high quantities of ATP to sustain axoplasmic transport, to maintain ionic gradients and membrane potential, and to conduct signal transduction along the axon and release and take up neurotransmitters at synaptic clefts. These characteristics contribute to the extreme dependence of neurons on “power” production by mitochondria. In addition to the extreme importance of energy supply for neurons, mitochondrial morphology differs in different parts of neurons. The mitochondrial network is elongated in dendrites and close to the cell body, but tends to be more fragmented in axons, probably to enhance transport of mitochondria along great distances. Moreover, mitochondria in neurons play an important role in regulating calcium homeostasis through their ability to take up calcium via several calcium uniporters, and release it back to the cytosol via several transporters. Synaptic mitochondria are always exposed to extensive Ca2+ influx and they can use the negative mitochondrial membrane potential created via the oxidative phosphorylation chain to buffer the cytosolic Ca2+ concentration. Furthermore, mitochondria have an average half-life of 10 to 25 days in neurons, longer than in other post-mitotic tissues, and in this regard are more likely to accumulate damage; thus, mitochondria in neurons are more vulnerable to I/R insult.
Mitochondria and Ischemia-Reperfusion (I/R) Injury
Recent publications indicated that mitochondria might play a key role in I/R injury. Mitochondria, also known as “power houses”, are highly abundant in neurons to maintain brain function. It is clear that mitochondria are dynamic organelles that frequently go through cycles of fusion and fission in a “kiss and run” pattern [5]. They continuously join by the process of fusion and divide by the process of fission, and the balance between these 2 processes plays a decisive role in the death and survival of cells, including neurons [6].
Mitochondrial fission and fusion processes are both mediated by large guanosine triphosphatases (GTPases) in the dynamin family that are well conserved in cells: dynamin-related protein 1 (Drp1) for fission, mitofusin1 (Mfn1) and mitofusin2 (Mfn2) for outer membrane fusion and Optic atrophy 1 (Opa1) for inner membrane fusion [7–9]. I/R can induce the late expression of these proteins, as well as post-transcriptional modifications, thereby causing brain dysfunction via interfering in the balance of fission and fusion. In this review we summarize recent research to help understand the signaling networks involved in the aberrant mitochondrial fusion and fission contributing to neuronal death caused by I/R injury, and the potential neuroprotective therapeutics targeting mitochondrial dynamics.
Mitochondrial Fusion and Fission
To meet cellular metabolic demands and respond to various external stresses and pathological changes, mitochondrial morphology changes dynamically as a result of the network of fusion and fission. External stimuli such as UV irradiation and oxygen deprivation can damage the mitochondria, which, however, can be eliminated by mitochondrial fission. Of note, even though mitochondrial fission is usually relevant to cell proliferation with an increase in the number of mitochondria, its contents are evenly distributed into daughter mitochondria [10,11]. Fission creates a greater number of fragmented mitochondria, whereas fusion facilitates the joining of different mitochondria to generate elongated mitochondria. Fusion promotes complementation between dysfunctional mitochondria. Mitochondrial DNA (mtDNA) is prone to mutation under stress, and damaged proteins easily accumulate in some older mitochondria. In this case, fusion rescues damaged mitochondria by allowing functional mitochondria to complement dysfunctional mitochondria via diffusion and sharing of components between organelles. Moreover, fusion allows for maintenance of a homogeneous mitochondrial network via mixing of mitochondrial contents between organelles [12]. It is generally agreed that fusion connects mitochondria to maximize the functional capacity, whereas fission separates mitochondria to eliminate “uncorrectable” ones. For neurons, mitochondrial distribution is highly dependent on these 2 competing yet interrelated processes [13]. Proper distribution of mitochondria is critical for axonal and synaptic function in peripheral neurons. However, aberrant mitochondrial fusion causing abnormal clustering of small fragmented mitochondria in neuronal cell bodies and proximal axons can significantly disrupt the transport of mitochondria in axons and lead to neurodegenerative disease [14]. In the cerebellum, Purkinje neurons are largely dependent on mitochondrial fusion to maintain correct localization of mitochondria in dendrites [15]. In hippocampal neurons, augmentation of mitochondrial fission causes fragmentation of the mitochondria and increases the abundance of dendritic mitochondria and the density of dendritic spines [16]. When these 2 processes occur correctly and are well coordinated, neurons perform perfectly.
Maintaining the Balance Between Mitochondrial Fusion and Fission
The balance between mitochondrial fission and mitochondrial fusion depends on proteins that are highly evolutionarily conserved. Mitochondria are enveloped by 2 closely apposed boundary membranes, and the fusion of these 2 membranes occurs independently [17]. On the outer mitochondrial membrane (OMM), MFN1 and MFN2 regulate the process of OMM fusion together. They both contain an amino-terminal GTP-binding domain, 2 coiled-coil domains, and a COOH-terminal with a bipartite transmembrane domain [18]. Furthermore, MFN2 is also involved in ER–mitochondria cross-talk, which is responsible for maintaining calcium homeostasis [19] and MFN1 is involved in an essential functional partner of Opa1 [19,20]. The ablation of either MFN1 or MFN2 in neurons induces a low rate of mitochondrial fusion; however, the expression levels of MFN1 and MFN2 are unequal, and neurons are more susceptible to mutations in the dominant protein [15]. In Purkinje cells (PCs), Mfn2-deficiency induces impaired respiratory complex activity and defects in inner membrane structure characteristic of respiratory dysfunction and eventually leads to cerebellar dysfunction [15]. Intriguingly, overexpression of MFN1 or MFN2 rescues PCs viability though increasing the total MFN complement and thus fusion activity [15]. Mitochondria without MFN2 expression are unable to either initiate or sustain fast processive movement and spend more time paused and undergo slower anterograde and retrograde movements [21], as explained by a previous study in which MFN2 knock-out mice had abnormal axonal mitochondrial distribution, with clustering of small fragmented mitochondria in neuronal cell bodies and proximal axons [14], disrupting the microtubule-based transport systems. Furthermore, mutation of MFN2 is the main pathogenesis of Charcot-Marie-Tooth (CMT) disease type 2A, which involves improper mitochondrial distribution and mitochondrial abnormalities in peripheral nerves [22]. Opa1 is localized in the inner mitochondrial membrane (IMM) and regulates IMM fusion. Opa1 expression goes through alternative splicing, as well as proteolytic processing, generating multiple variant forms that include IMM-anchored long Opa1 (L-Opa1) and soluble short Opa1 (S-Opa1), which play different roles in fusion [23]. Moreover, Opa1 has been shown to be involved in cristae integrity and mtDNA maintenance [24]. Inhibition of mitochondrial fusion via interfering RNA-mediated knockdown of Opa1 can lead to reduced expression of pre- and post-synaptic marker proteins and a decrease in synapse numbers in the early period, and with extended culturing, dendritic growth becomes affected, and synaptic protein expression remains low [25].
The most investigated proteins involved in mitochondrial fission are the Drp1 and fission protein 1 (Fis1). The localization of Drp1 is mainly cytosolic, but a fraction of this protein is found on the OMM. Drp1 generally has 4 important domains: GTPase domain, the middle domain, the GTPase effector domain (GED), and the variable domain [26,27]. The GED domain is important for stimulation of GTPase activity, formation and stability of higher-order complexes, and efficient mitochondrial division. Moreover, the fission-promoting activity of Drp1 is controlled by many post-translational modifications around the variable domain, including phosphorylation, SUMOylation, ubiquitination and S-nitrosylation [28]. It had been reported that NS-Drp1−/− mice (generated using a transgenic line) died on the first day after parturition, and analysis of serial sections of E18.5 embryos revealed a reduction in size of the forebrain, with expanded subdural space and ventricles [29]. It has also been found that primary culture of NS-Drp1−/− mouse forebrain showed a decreased number of neurites and defective synapse formation [29]. For the development of cerebella, compared to granule cells, mitochondria in En1-Drp1KO Purkinje cells had lower quantities and large round structures [30]. Moreover, by visual inspection, the cerebella of En1-Drp1KO mice had completely smooth surfaces, their size was decreased to 60% of control cerebella, and lobule fissures were barely detectable [30]. Inhibition of mitochondrial fission by knockdown of Drp1 leads to clustering of mitochondria in the cell body and when Drp1 synapses are stimulated at high frequency [31]. Berthet et al. also found that in axons of Drp1 knock-out dopamine (DA) neurons, mitochondria were characterized by impaired coordination of mitochondrial movements and decreased overall mass [32]. Furthermore,
Dysfunctional Mitochondria in Neurons During the I/R Process
Cerebral I/R injury promotes changes in biological behavior of mitochondria, which can lead to mitochondrial dysfunction [36,37]. I/R can induce a “burst” of ROS generation that promotes the release of cytochrome C and triggers the neuronal apoptosis signaling pathway [38]. Furthermore, I/R leads to destruction of mitochondrial membrane potential, which plays an important role in ROS generation. Moreover, during the I/R pathological process, intracellular Ca2+ homeostasis is interrupted through the opening of mitochondrial permeability transition pores, and the overloaded Ca2+ can cause mitochondrial swelling [38]. All of these changes mentioned above result in significant changes in mitochondrial morphology, alter the mitochondrial dynamics, and induce neuronal death. Therefore, mitochondrial dysfunction occurs during the whole process of I/R injury, causing a vicious cycle and leading to mitochondrial-mediated cell death.
Unbalanced Mitochondrial Fusion/Fission Process Leads to Neuronal Apoptosis
The balance of mitochondrial fusion and fission is necessary to maintain mitochondrial integrity, which is involved in regulating neuronal death [39]. Increasing evidence indicates that I/R can induce mitochondrial fusion and fission abnormalities, consequently resulting in neuronal death (Table 1) [40–46]. Evidence suggests that downregulation of fusion proteins enhance I/R-induced neuronal apoptosis, while upregulation of fusion proteins suppresses I/R-induced neuronal apoptosis by facilitating mitochondrial fusion. It had been reported that the number of Opa1-positive cells increased over time in the ischemic penumbra after 90 min of tMCAO and reached a peak at 2 days after reperfusion, which suggests activation of mitochondrial fusion for cell survival in the penumbra [47]. However, in the ischemic core part, western blot analysis showed a clear decrease of Opa1 protein levels at 90 min of MCAO and 2 days of reperfusion, suggesting that the necrotic brain damage in the core part is irreversible [47]. Under I/R condition, upregulation Opa1 facilitates mitochondrial fusion, reverses the mitochondrial interconnective morphology, and alleviates I/R-induced neuronal apoptosis, reducing the volume of infarction [48]. Mild Opa1 overexpression is compatible with life and protects mice from I/R injury via cristae remodeling in brain infarction [49]. Moreover, I/R also changes the amount of mitofusin1/2, which affects the process of mitochondrial fusion. The MFN1 protein expression increased immediately and continued for 24 h of reoxygenation [50], whereas the expression of MFN2 mRNA and protein were mildly elevated in 24 h after reperfusion, then markedly decreased after 36 h and 48 h [51]. Moreover, by regulating the expression of MFN2, overexpression of MFN2 significantly increased neuronal viability and decreased cleaved caspase 3 levels, which plays an important role in cell apoptosis [51]. MFN2 overexpression promotes mitochondrial fusion to inhibit hypoxia-induced apoptosis through attenuated mitochondrial dysfunction and restored mitochondrial morphology; however, depletion of MFN2 can lead to apoptosis in normal and hypoxic conditions [52]. The above evidence suggests that enhancement of mitochondrial fusion may have protective effects on neurons after I/R injury. However, Sanderson et al reported the opposite results regarding alteration of mitochondrial fusion proteins induced by I/R injury. They found that upon I/R, multiple changes in Opa1 occur, including: (i) translocation of Opa1 from the mitochondria to the cytosol; (ii) increase in the short isoform of Opa1, suggestive of proteolytic processing; and (iii) breakdown of Opa1 oligomers in the mitochondria [53]. These changes eventually lead to a significant reduction in the number of Opa1 in the mitochondria and irresistible fission, causing a series of problems such as disruption of the IMM structure, dissipation of the MM potential, release of CytC, impaired Ca2+ homeostasis, and even neuronal apoptosis [54]. Under I/R conditions, the changes in mitochondrial fusion proteins are widely divergent in various studies. We consider that this difference may be related to the following reasons. Whether
Several studies have suggested that mitochondrial fission plays a key role in regulating neuronal apoptosis under I/R condition. Liu et al. found that Drp1 and phosphorylation of Drp1 site Ser637 (P-Drp1) were both upregulated after tMCAO, with a peak at 2 and 14 days, respectively, suggesting an increase in mitochondrial fission in I/R condition [47]. In addition, translocation of Drp-1 from cytoplasm to mitochondrial membrane was promoted under I/R condition [56]. Moreover, knockdown or inhibition of Drp1 expression by Drp1 inhibitor (MDIVI-1) and Drp1-siRNA can suppress mitochondrial fission, inhibit neuronal apoptosis, reduce brain infarct volume, and attenuate I/R-induced cerebral injury [57]. The above evidence suggests that stimulating mitochondrial fission via overexpression of Drp1 may have a detrimental effect on neurons after I/R attack. However, Zuo et al. found the opposite effects of Drp1 on neuronal apoptosis induced by I/R injury [56]. Neurons with deficient Drp1, which is silenced or inhibited by siRNA or pharmacological inhibitor MDIVI-1, are more vulnerable in I/R [56]. This indicates Drp1 may play a protective role in neuronal apoptosis. Further analysis suggests that disturbed mitochondrial fission by inhibiting Drp1 contributes to interruption of mitochondrial self-purification, and accumulated damaged to mitochondria could lead to mitochondria-related injury after I/R [58]. The dramatically opposed conclusions suggest that mitochondrial fission may have pro-apoptosis effects and anti-apoptosis effects on neurons in I/R conditions. Determining which effect has the major role in neuronal apoptosis induced by I/R is very important for understanding the pathogenesis of disease.
Mitochondrial Autophagy Is Involved in Neuronal Death During I/R
Autophagy is proved to be involved in I/R. In most situations, autophagy plays a protective role under I/R conditions by cleaning damaged organelles and proteins [59]. However in the late I/R period, the grossly enhanced autophagy can contribute to autophagic cell death, leading to subsequent damage of neurons [60]. Mitophagy is a process of selective mitochondrial autophagy, which can clean damaged mitochondria selectively in cells and plays a protective role in neuronal death induced by I/R injury. In response to I/R injury, it was observed that mitochondria swelled after 6-h treatment with I/R and cristae disrupted after 12 h, while after 24 h, autophagosomes, mitophagy, and partially degraded mitochondria were observed in neurons [61]. Therefore, it is clear that there exists neuronal mitophagy under I/R conditions. Mitochondrial fusion/fission is still activated in ischemia penumbra in order to promote mitophagy and neuronal survival [62]. Fission can generate normal mitochondria, which tend to fuse together, while fission also can generate dysfunctional mitochondrial with decreased membrane potential, which are degraded by mitophagy. Moreover, excessive fusion of mitochondria has been proved to inhibit the mitophagy process [63]. There is speculation that fragmented mitochondria are more easily enwrapped by autophagosomes compared to larger elongated mitochondria [64]. The above findings suggest that mitophagy is promoted more by fission than by fusion. However, the underlying mechanisms by which mitochondrial fission promotes mitophagy in I/R are unclear.
Mitochondrial Dynamics and Other Forms of Neuronal Death Induced by I/R
I/R injury can trigger an outbreak of inflammation in the lesions and lead to activation of Caspase-1, resulting in inhibition of mitophagy and excessive fission of mitochondria, thereby inducing neuronal pyroptosis and promoting neuronal death [65]. Moreover, I/R injury can induce neuronal ferroptosis, accompanied by loss of mitochondrial membrane potential and enhanced mitochondrial fragmentation, which may be involved in excessive mitochondrial fission [66].
Molecular Mechanisms Underlying Dysfunctional Mitochondrial Dynamics During the I/R Process
Numerous studies have shown that aberrant mitochondrial fusion/fission induced by I/R is the key component that causes neuronal cell death. Here, we summarize the underlying mechanisms by which I/R influences the processes of mitochondrial fusion and fission in neurons.
Mitochondrial fission is mainly mediated by Drp1, which is mainly localized in the cytosol and is recruited to the outer mitochondrial membrane with the help of Fis. During the I/R process, Drp1 is considered a key regulatory protein that causes an exaggerated fission process and leads to mitochondria-mediated cell death. When the intracellular environment changes, Drp1 is post-translationally modified by SUMOylation/deSUMOylation, S-nitrosylation, or phosphorylation/dephosphorylation of its variable domain [67]. In these processes, SUMOylation/deSUMOylation and phosphorylation/dephosphorylation are considered to be involved in I/R-induced Drp1 activation and neuronal apoptosis.
In humans, Drp1-dependent mitochondrial fission is controlled by phosphorylation at 4 different conserved sites: Ser637, Ser616 (equivalent to Ser585 of rat Drp1), Ser656, and Ser693 [68,69]. Ser637 is the first identified serine residue site that is phosphorylated by protein kinase A (PKA) and is dephosphorylated by Ca2+-dependent phosphatase calcineurin (CaN) [70]. Cdk1/cyclin B is involved in the phosphorylation of Ser616 (equivalent to Ser585 of rat Drp1) [69]. Similar to Ser637, Ser 656 is phosphorylated by cyclic AMP-dependent protein kinase and dephosphorylated by calcineurin and PP2A/Bβ2 [71]. Moreover, GSK3β can regulate the phosphorylation of Ser693. I/R can induce an increase in the phosphorylation of Drp1 at serine 616 via activation of Cdk1/cyclin B, thereby stimulating Drp1 mitochondrial recruitment and neuronal mitochondrial fission, without significantly affecting the expression of total Drp1 proteins [72]. Chen et al found an antagonistic mechanism by which I/R can increase PINK1 expression, causing downregulation of Drp1 phosphorylation at serine 616, which plays a protective role in neuronal apoptosis [72]. Moreover, the elevated level of PINK1 after I/R injury facilitates mitophagy through the PINK1/Parkin/p62 pathway, moderating neuronal death [61]. I/R can also suppress the volume of p-Drp1 (Ser637), and Ser637 tends to be dephosphorylated by activation of calcineurin to increase Drp1 activity and promote mitochondrial fission [73]. CaN-mediated Drp1 dephosphorylation at Ser656 also contributes to neuronal death via stimulating neuronal mitochondrial fission during I/R [74] (Figure 1).
SUMOylation plays an important role in a wide range of cellular processes. There are 3 SUMO paralogues (SUMO-1/3) in mammals that are conjugated to lysine residues in target proteins [75]. SUMO-2/3 share only approximately 50% sequence similarity with SUMO-1, but SUMO-2 and -3 differ by just 3 N-terminal amino acids and are often referred to collectively as SUMO-2/3 [75]. SUMOylation is a dynamic process that is readily reversed by a family of sentrin/SUMO-specific proteases (SENP) in humans [76]. The nuanced SUMO-mediated regulation system is involved in mitochondrial dynamics, as recent evidence suggests that Drp1 SUMOylation by SUMO-1 or deSUMOylation by SENPs can facilitate its localization in mitochondria, promoting mitochondrial fission, whereas Drp1 SUMOylation by SUMO-2/3 prevents it [77]. During ischemia, SENP3 is selectively degraded to increase Drp1 SUMO-2/3-ylation and inhibit its localization to the mitochondria, which contributes to reduced mitochondrial fission and neuronal apoptosis. However, during reoxygenation after OGD, SENP3 levels are restored, leading to the recruitment of Drp1 and increased mitochondrial fission, thereby increasing neuronal apoptosis [77] (Figure 2).
Besides the over-activated mitochondrial fission, I/R can also decrease Opa1 levels to influence the process of mitochondrial fusion. Opa1 has 8 isoforms which generally contain S1 and S2 cleavage sites, or an S1 site alone [78]. Cleavage at S2 is regulated by i-AAA protease Yme1L, which produces an appropriate level of long and short Opa1 products, optimal for Opa1 functioning [79]. When mitochondria lose membrane potential, Opa1 tends to be cut by OMA1, a zinc-metalloprotease which resides on the inner membrane, at the S1 site inducing aberrant mitochondrial fusion [80]. I/R induces a discernible decrease in Yme1L protein expression, whereas OMA1 is not altered significantly, resulting in an imbalance between long (L-Opa1) and short (S-Opa1) forms (the level of L-Opa1 is less than S-Opa1), which may favor mitochondrial fission and promote neuronal apoptosis [81]. Moreover, I/R injury can downregulate Mfn2 expression, then disrupt Mfn2-mediated mitophagy through the MAPK–ERK–CREB signaling pathway, thereby suppressing the protective effect of mitophagy in neurons [82].
Experimental Interventions Targeting Dysfunctional Mitochondrial Dynamics Induced by I/R
There is sufficient evidence to prove that I/R causes aberrant mitochondrial fusion and fission, resulting in neuronal damage. Therefore, restoring the balance between mitochondrial fusion and fission may be a new target for the treatment of I/R-induced neuronal injury. Genetic interventions or pharmacological agents that either inhibit fission or facilitate fusion generally protect neurons from I/R injury.
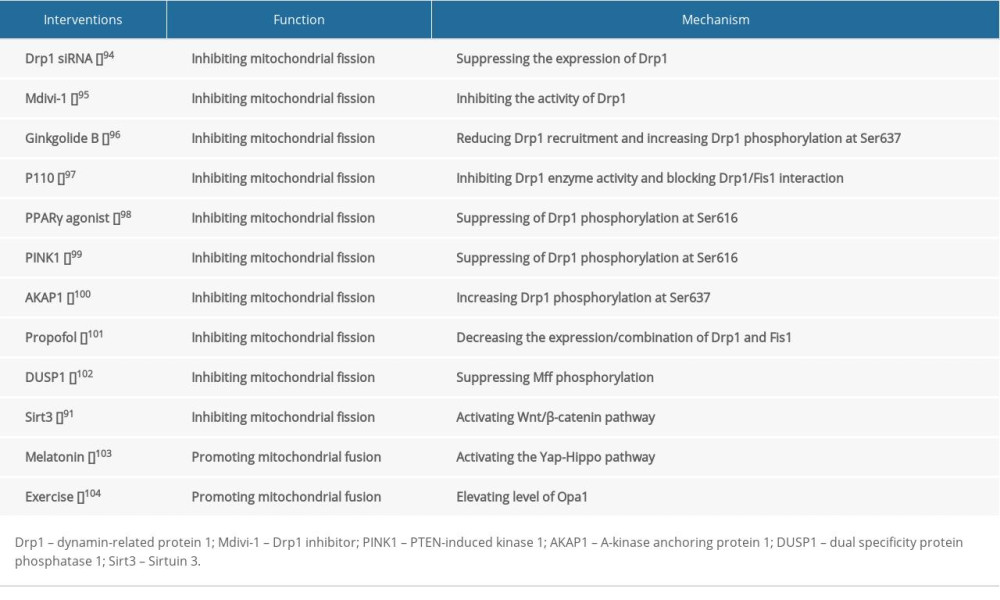
Intervention with the pharmacological inhibitor of Drp1 (mdivi-1) or Drp1 siRNA can suppress excessive mitochondrial fission, maintain mitochondrial membrane potential, and inhibit cell death after I/R-induced brain injury
Several drugs had been clinically used to treat cerebral I/R injury. Propofol can inhibit mitochondrial fission by decreasing the expression of Drp1, as well as the combination of Drp1 and Fis1 [89]. DUSP1 overexpression suppresses Mff phosphorylation via JNK-Mff pathways, thereby inhibiting excessive mitochondrial fission and attenuating neuronal death caused by I/R injury [90]. Sirtuin 3 (Sirt3) blocks mitochondrial fission and induces pro-survival signals in neurons subjected by I/R through activating the Wnt/β-catenin pathway [91]. Promoting mitochondrial fusion also exerts neuro-protective effects. Mfn2 overexpression maintains mitochondrial morphology and attenuates I/R-induced apoptosis [92]. Melatonin supplementation promotes Opa1-meditated mitochondrial fusion via activating the Yap-Hippo pathway, ultimately reducing brain reperfusion damage [48]. Interestingly, rats pretreated with treadmill training have higher levels of Opa1and are less vulnerable to I/R stress, suggesting that moderate exercise before I/R injury can protect the brain [93–104] (Table 2).
Cerebral Ischemia-Reperfusion Drugs Targeting Mitochondrial Dysfunction
Cerebral ischemia-reperfusion treatments include ischemic preconditioning, ischemic postconditioning, pharmacological preconditioning, and drug postconditioning. However, their clinical efficacy is not satisfactory. In recent years, researchers have found that mitochondrial dysfunction was one of the main factors in I/R, and mitochondria are an attractive target in I/R neuroprotection. Therefore, mitochondrial-targeted therapy of the nervous system for I/R gradually starts from basic study to clinical application. Mitochondrial-targeted therapy is a kind of therapy for specific parts of damaged mitochondria, which can counteract the adverse events caused by mitochondrial dysfunction. Through mitochondrial-targeted therapy, targeted drugs are given to inhibit the adverse events caused by mitochondrial dysfunction, make it return to normal from a pathological state, and reduce mitochondrial damage, so as to improve neuronal cell viability and inhibit I/R injury.
Drugs Targeting Mitochondrial ATP Channel
Nicorandil is a sensitive opener of the mitochondrion KATP channel. By activating the KATP channel, nicorandil makes a large amount of K+ enter into mitochondria and activate ETC [105]. ATP synthesis increases while inhibiting ATP consumption and preserving ATP, thus reducing the damage range of nerve cells. At the same time, nicorandil can also reduce Ca2+ overload, inhibit ROS burst, and protect nerve cells [106].
Drugs Targeting Mitochondrial Ca2
Mitochondrial calcium uniporter (MUC) is a protein located in the inner membrane of mitochondria, which is responsible for the absorption of mitochondrial Ca2+. MCU inhibitors can reduce the expression of MCU, reduce the content of mitochondrial Ca2+, and weaken Ca2+-dependent apoptosis. RUR and RU360 have been widely used as inhibitors of MCU [107]. Sodium calcium exchanger (NCX) reverse transport inhibitors and Na+/H+ exchanger (NHE) inhibitors also have good effects on reducing Ca2+ overload. Rapamycin can reduce Ca2+ concentration and resist I/R injury by activating the positive mode of NCX. NHE inhibitors such as KR-32570 and HOE694 can activate the positive mode of NCX and reduce the concentration of Ca2+ by inhibiting the accumulation of Na + in cells, thus exerting neuroprotective effects [108].
Conclusions
The present review summarizes recent research results on the role of mitochondria in I/R injury and clarifies the complex mechanism of mitochondrial fission and fusion as related to neuronal apoptosis under I/R conditions. We also addressed the anti-cerebral ischemia-reperfusion drugs targeting mitochondrial dysfunction. Tremendous progress has been made in basic research toward novel treatment of I/R injury, but therapies that have been effective to I/R patients still are few and far from clinical application. As mentioned above, it is still unclear whether fusion and fission play pro-apoptosis roles during cerebral I/R injury. Moreover, the non-fusion/fission effects of mitochondrial fusion/fission proteins are unclear and the requirement for balance between mitochondrial fusion and fission is rigorous. Further research is needed to elucidate the molecular mechanism by which I/R regulates mitochondrial fusion and fission in neurons.
Figures
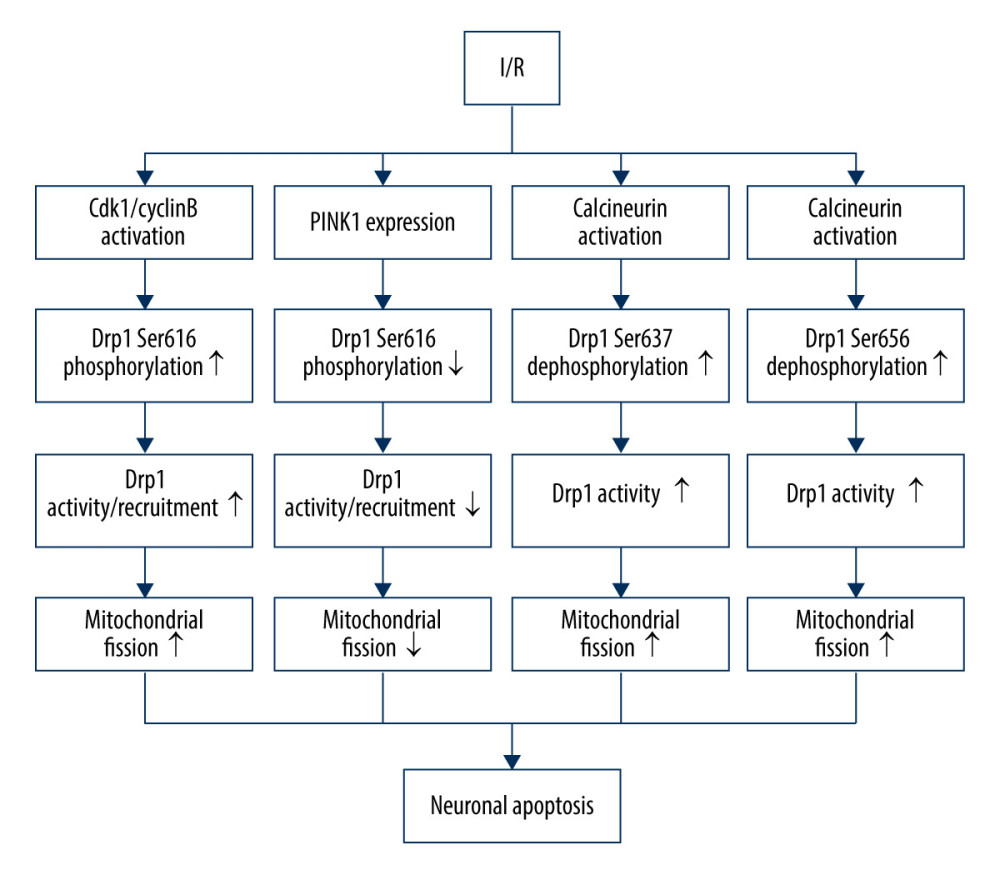 Figure 1. Drp1 phosphorylation/dephosphorylation and mitochondrial fission in neurons upon I/R. I/R activates Cdk1/cyclin B to induce the phosphorylation of Drp1 at serine 616, leading to mitochondrial fission via and promoting the activation/recruitment of Drp1. I/R promotes PINK1 expression to suppress the Drp1 phosphorylation at serine 616, suppressing the activation/recruitment of Drp1 and mitochondrial fission. I/R activates calcineurin, leading to Drp1 Ser637/656 dephosphorylation, increasing Drp1 activity and promoting mitochondrial fission.
Figure 1. Drp1 phosphorylation/dephosphorylation and mitochondrial fission in neurons upon I/R. I/R activates Cdk1/cyclin B to induce the phosphorylation of Drp1 at serine 616, leading to mitochondrial fission via and promoting the activation/recruitment of Drp1. I/R promotes PINK1 expression to suppress the Drp1 phosphorylation at serine 616, suppressing the activation/recruitment of Drp1 and mitochondrial fission. I/R activates calcineurin, leading to Drp1 Ser637/656 dephosphorylation, increasing Drp1 activity and promoting mitochondrial fission. 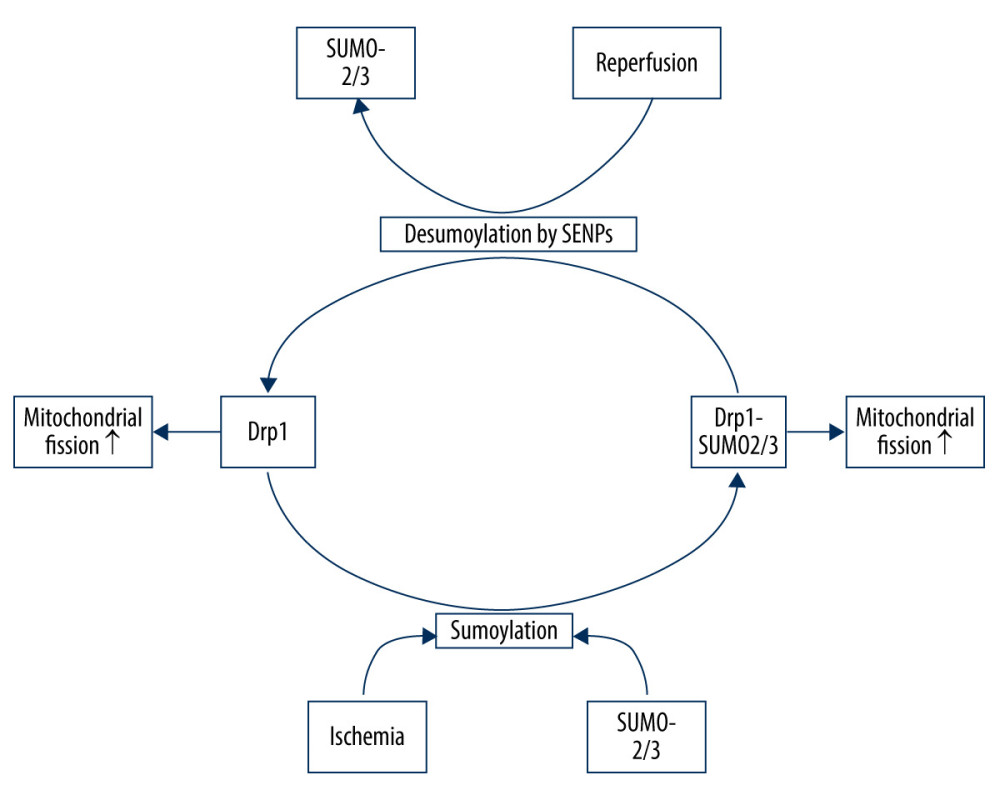 Figure 2. Drp1 SUMOylation/deSUMOylation and mitochondrial fission in neurons upon I/R. During ischemia, Drp1 SUMOylation by SUMO-2/3 contributes to reduced mitochondrial fission; however, after reperfusion, SENP3 levels are restored, leading to the deSUMOylation of Drp1-SUMO2/3 and increased mitochondrial fission.
Figure 2. Drp1 SUMOylation/deSUMOylation and mitochondrial fission in neurons upon I/R. During ischemia, Drp1 SUMOylation by SUMO-2/3 contributes to reduced mitochondrial fission; however, after reperfusion, SENP3 levels are restored, leading to the deSUMOylation of Drp1-SUMO2/3 and increased mitochondrial fission. 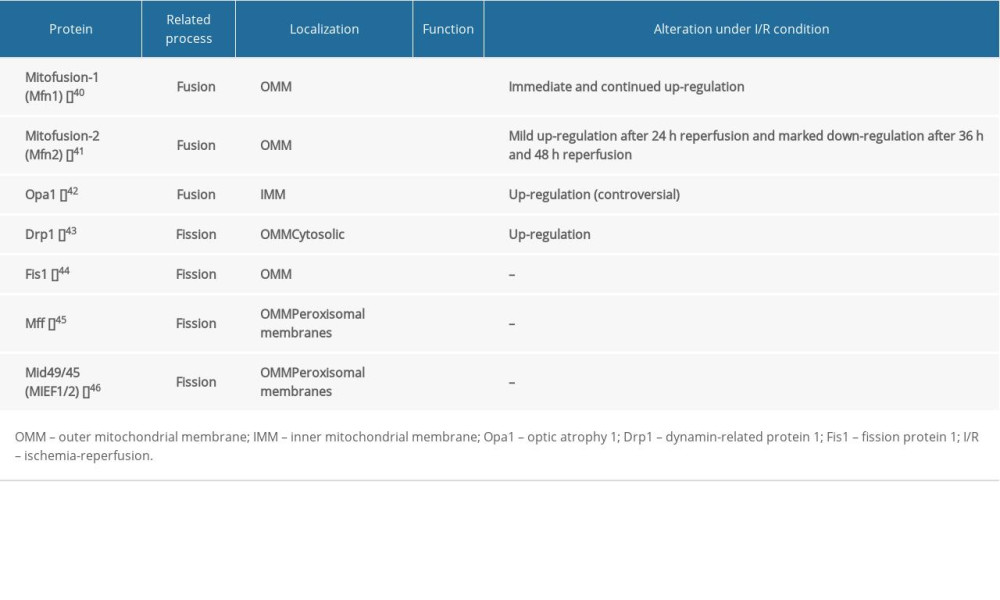
References
1. Kalogeris T, Baines CP, Krenz M, Korthuis RJ, Cell biology of ischemia/reperfusion injury: Int Rev Cell Mol Biol, 2012; 298; 229-317
2. Sharma D, Maslov LN, Singh N, Jaggi AS, Remote ischemic preconditioning-induced neuroprotection in cerebral ischemia-reperfusion injury: Preclinical evidence and mechanisms: Eur J Pharmacol, 2020; 883; 173380
3. Tilokani L, Nagashima S, Paupe V, Prudent J, Mitochondrial dynamics: Overview of molecular mechanisms: Essays Biochem, 2018; 62; 341-60
4. Kim J, Cheong JH, Role of mitochondria-cytoskeleton interactions in the regulation of mitochondrial structure and function in cancer stem cells: Cells, 2020; 9; 1691
5. Viana MP, Brown AI, Mueller IA, Mitochondrial fission and fusion dynamics generate efficient, robust, and evenly distributed network topologies in budding yeast cells: Cell Syst, 2020; 10; 287-97.e5
6. Fu W, Liu Y, Yin H, Mitochondrial dynamics: biogenesis, fission, fusion, and mitophagy in the regulation of stem cell behaviors: Stem Cells Int, 2019; 2019 9757201
7. Meeusen S, DeVay R, Block J, Mitochondrial inner-membrane fusion and crista maintenance requires the dynamin-related GTPase Mgm1: Cell, 2006; 127; 383-95
8. Song Z, Ghochani M, McCaffery JM, Mitofusins and OPA1 mediate sequential steps in mitochondrial membrane fusion: Mol Biol Cell, 2009; 20; 3525-32
9. Yu F, Abdelwahid E, Xu T, The role of mitochondrial fusion and fission in the process of cardiac oxidative stress: Histol Histopathol, 2020; 35; 541-52
10. Parone PA, Da Cruz S, Tondera D, Preventing mitochondrial fission impairs mitochondrial function and leads to loss of mitochondrial DNA: PLoS One, 2008; 3; e3257
11. Möpert K, Hajek P, Frank S, Loss of Drp1 function alters OPA1 processing and changes mitochondrial membrane organization: Exp Cell Res, 2009; 315; 2165-80
12. Chen H, Chomyn A, Chan DC, Disruption of fusion results in mitochondrial heterogeneity and dysfunction: J Biol Chem, 2005; 280; 26185-92
13. Detmer SA, Chan DC, Functions and dysfunctions of mitochondrial dynamics: Nat Rev Mol Cell Biol, 2007; 8; 870-79
14. Baloh RH, Schmidt RE, Pestronk A, Milbrandt J, Altered axonal mitochondrial transport in the pathogenesis of Charcot-Marie-Tooth disease from mitofusin 2 mutations: J Neurosci, 2007; 27; 422-30
15. Chen H, McCaffery JM, Chan DC, Mitochondrial fusion protects against neurodegeneration in the cerebellum: Cell, 2007; 130; 548-62
16. Li Z, Okamoto K, Hayashi Y, Sheng M, The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses: Cell, 2004; 119; 873-87
17. Malka F, Guillery O, Cifuentes-Diaz C, Separate fusion of outer and inner mitochondrial membranes: EMBO Rep, 2005; 6; 853-59
18. Farmer T, Naslavsky N, Caplan S, Tying trafficking to fusion and fission at the mighty mitochondria: Traffic, 2018; 19; 569-77
19. Cipolat S, Martins de Brito O, Dal Zilio B, Scorrano L, OPA1 requires mitofusin 1 to promote mitochondrial fusion: Proc Natl Acad Sci USA, 2004; 101; 15927-32
20. Naon D, Zaninello M, Giacomello M, Critical reappraisal confirms that Mitofusin 2 is an endoplasmic reticulum-mitochondria tether: Proc Natl Acad Sci USA, 2016; 113; 11249-54
21. Misko A, Jiang S, Wegorzewska I, Mitofusin 2 is necessary for transport of axonal mitochondria and interacts with the Miro/Milton complex: J Neurosci, 2010; 30; 4232-40
22. Detmer SA, Vande Velde C, Cleveland DW, Chan DC, Hindlimb gait defects due to motor axon loss and reduced distal muscles in a transgenic mouse model of Charcot-Marie-Tooth type 2A: Hum Mol Genet, 2008; 17; 367-75
23. Ishihara N, Fujita Y, Oka T, Mihara K, Regulation of mitochondrial morphology through proteolytic cleavage of OPA1: EMBO J, 2006; 25; 2966-77
24. Del Dotto V, Mishra P, Vidoni S, OPA1 Isoforms in the Hierarchical Organization of Mitochondrial Functions: Cell Rep, 2017; 19; 2557-71
25. Bertholet AM, Millet AM, Guillermin O: Brain, 2013; 136; 1518-33
26. Smirnova E, Shurland DL, Ryazantsev SN, van der Bliek AM, A human dynamin-related protein controls the distribution of mitochondria: J Cell Biol, 1998; 143; 351-58
27. Okamoto K, Shaw JM, Mitochondrial morphology and dynamics in yeast and multicellular eukaryotes: Annu Rev Genet, 2005; 39; 503-36
28. Cho B, Choi SY, Cho HM, Physiological and pathological significance of dynamin-related protein 1 (drp1)-dependent mitochondrial fission in the nervous system: Exp Neurobiol, 2013; 22; 149-57
29. Ishihara N, Nomura M, Jofuku A, Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice: Nat Cell Biol, 2009; 11; 958-66
30. Wakabayashi J, Zhang Z, Wakabayashi N, The dynamin-related GTPase Drp1 is required for embryonic and brain development in mice: J Cell Biol, 2009; 186; 805-16
31. Verstreken P, Ly CV, Venken KJ, Synaptic mitochondria are critical for mobilization of reserve pool vesicles at Drosophila neuromuscular junctions: Neuron, 2005; 47; 365-78
32. Berthet A, Margolis EB, Zhang J, Loss of mitochondrial fission depletes axonal mitochondria in midbrain dopamine neurons: J Neurosci, 2014; 34; 14304-17
33. Yoon Y, Krueger EW, Oswald BJ, McNiven MA, The mitochondrial protein hFis1 regulates mitochondrial fission in mammalian cells through an interaction with the dynamin-like protein DLP1: Mol Cell Biol, 2003; 23; 5409-20
34. Palmer CS, Elgass KD, Parton RG, Adaptor proteins MiD49 and MiD51 can act independently of Mff and Fis1 in Drp1 recruitment and are specific for mitochondrial fission: J Biol Chem, 2013; 288; 27584-93
35. Osellame LD, Singh AP, Stroud DA, Cooperative and independent roles of the Drp1 adaptors Mff, MiD49 and MiD51 in mitochondrial fission: J Cell Sci, 2016; 129; 2170-81
36. Li J, Ma X, Yu W, Reperfusion promotes mitochondrial dysfunction following focal cerebral ischemia in rats: PLoS One, 2012; 7; e46498
37. Sun J, Li YZ, Ding YH: Brain Res, 2014; 1589; 126-39
38. Hu Y, Deng H, Xu S, Zhang J, MicroRNAs regulate mitochondrial function in cerebral ischemia-reperfusion injury: Int J Mol Sci, 2015; 16; 24895-917
39. Suen DF, Norris KL, Youle RJ, Mitochondrial dynamics and apoptosis: Genes Dev, 2008; 22; 1577-90
40. Chung SH, Calafiore M, Plane JM, Apoptosis inducing factor deficiency causes reduced mitofusion 1 expression and patterned Purkinje cell degeneration: Neurobiol Dis, 2011; 41; 445-57
41. Sood A, Jeyaraju DV, Prudent J, A Mitofusin-2-dependent inactivating cleavage of Opa1 links changes in mitochondria cristae and ER contacts in the postprandial liver: Proc Natl Acad Sci USA, 2014; 111; 16017-22
42. Belenguer P, Pellegrini L, The dynamin GTPase OPA1: More than mitochondria: Biochim Biophys Acta, 2013; 1833; 176-83
43. Zhou BH, Wei SS, Jia LS, Drp1/Mff signaling pathway is involved in fluoride-induced abnormal fission of hepatocyte mitochondria in mice: Sci Total Environ, 2020; 725; 138192
44. Scarpelli PH, Tessarin-Almeida G, Viçoso KL, Melatonin activates FIS1, DYN1, and DYN2 Plasmodium falciparum related-genes for mitochondria fission: Mitoemerald-GFP as a tool to visualize mitochondria structure: J Pineal Res, 2019; 66; e12484
45. Lewis TL, Kwon SK, Lee A, MFF-dependent mitochondrial fission regulates presynaptic release and axon branching by limiting axonal mitochondria size: Nat Commun, 2018; 9; 5008
46. Yu R, Liu T, Jin SB, MIEF1/2 function as adaptors to recruit Drp1 to mitochondria and regulate the association of Drp1 with Mff: Sci Rep, 2017; 7; 880
47. Liu W, Tian F, Kurata T, Dynamic changes of mitochondrial fusion and fission proteins after transient cerebral ischemia in mice: J Neurosci Res, 2012; 90; 1183-89
48. Wei N, Pu Y, Yang Z, Therapeutic effects of melatonin on cerebral ischemia reperfusion injury: Role of Yap-OPA1 signaling pathway and mitochondrial fusion: Biomed Pharmacother, 2019; 110; 203-12
49. Varanita T, Soriano ME, Romanello V, The OPA1-dependent mitochondrial cristae remodeling pathway controls atrophic, apoptotic, and ischemic tissue damage: Cell Metab, 2015; 21; 834-44
50. Wappler EA, Institoris A, Dutta S, Mitochondrial dynamics associated with oxygen-glucose deprivation in rat primary neuronal cultures: PLoS One, 2013; 8; e63206
51. Peng C, Rao W, Zhang L, Mitofusin 2 exerts a protective role in ischemia reperfusion injury through increasing autophagy: Cell Physiol Biochem, 2018; 46; 2311-24
52. Peng C, Rao W, Zhang L, Mitofusin 2 ameliorates hypoxia-induced apoptosis via mitochondrial function and signaling pathways: Int J Biochem Cell Biol, 2015; 69; 29-40
53. Sanderson TH, Raghunayakula S, Kumar R, Neuronal hypoxia disrupts mitochondrial fusion: Neuroscience, 2015; 301; 71-78
54. Kushnareva YE, Gerencser AA, Bossy B, Loss of OPA1 disturbs cellular calcium homeostasis and sensitizes for excitotoxicity: Cell Death Differ, 2013; 20; 353-65
55. Ong SB, Kalkhoran SB, Hernández-Reséndiz S, Mitochondrial-shaping proteins in cardiac health and disease – the long and the short of it: Cardiovasc Drugs Ther, 2017; 31; 87-107
56. Zuo W, Yang PF, Chen J, Drp-1, a potential therapeutic target for brain ischaemic stroke: Br J Pharmacol, 2016; 173; 1665-77
57. Chuang YC, Lin TK, Yang DI, Peroxisome proliferator-activated receptor-gamma dependent pathway reduces the phosphorylation of dynamin-related protein 1 and ameliorates hippocampal injury induced by global ischemia in rats: J Biomed Sci, 2016; 23; 44
58. Zuo W, Zhang S, Xia CY, Mitochondria autophagy is induced after hypoxic/ischemic stress in a Drp1 dependent manner: The role of inhibition of Drp1 in ischemic brain damage: Neuropharmacology, 2014; 86; 103-15
59. Su J, Zhang T, Wang K, Autophagy activation contributes to the neuroprotection of remote ischemic perconditioning against focal cerebral ischemia in rats: Neurochem Res, 2014; 39; 2068-77
60. Shui M, Liu X, Zhu Y, Wang Y, Exogenous hydrogen sulfide attenuates cerebral ischemia-reperfusion injury by inhibiting autophagy in mice: Can J Physiol Pharmacol, 2016; 94; 1187-92
61. Lan R, Wu JT, Wu T, Mitophagy is activated in brain damage induced by cerebral ischemia and reperfusion via the PINK1/Parkin/p62 signalling pathway: Brain Res Bull, 2018; 142; 63-77
62. Liu K, Sun Y, Gu Z, Mitophagy in ischaemia/reperfusion induced cerebral injury: Neurochem Res, 2013; 38; 1295-300
63. Twig G, Shirihai OS, The interplay between mitochondrial dynamics and mitophagy: Antioxid Redox Signal, 2011; 14; 1939-51
64. Ni HM, Williams JA, Ding WX, Mitochondrial dynamics and mitochondrial quality control: Redox Biol, 2015; 4; 6-13
65. Yu J, Nagasu H, Murakami T, Inflammasome activation leads to Caspase-1-dependent mitochondrial damage and block of mitophagy: Proc Natl Acad Sci USA, 2014; 111; 15514-19
66. Stockwell BR, Friedmann Angeli JP, Bayir H, A regulated cell death nexus linking metabolism, redox biology, and disease: Cell, 2017; 171; 273-85
67. Chang CR, Blackstone C, Dynamic regulation of mitochondrial fission through modification of the dynamin-related protein Drp1: Ann NY Acad Sci, 2010; 1201; 34-39
68. Cribbs JT, Strack S, Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death: EMBO Rep, 2007; 8; 939-44
69. Taguchi N, Ishihara N, Jofuku A, Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission: J Biol Chem, 2007; 282; 11521-29
70. Cereghetti GM, Stangherlin A, Martins de Brito O, Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria: Proc Natl Acad Sci USA, 2008; 105; 15803-8
71. Merrill RA, Slupe AM, Strack S, N-terminal phosphorylation of protein phosphatase 2A/Bβ2 regulates translocation to mitochondria, dynamin-related protein 1 dephosphorylation, and neuronal survival: FEBS J, 2013; 280; 662-73
72. Chen SD, Lin TK, Yang DI, Roles of PTEN-induced putative kinase 1 and dynamin-related protein 1 in transient global ischemia-induced hippocampal neuronal injury: Biochem Biophys Res Commun, 2015; 460; 397-403
73. Zhou X, Wang HY, Wu B, Ginkgolide K attenuates neuronal injury after ischemic stroke by inhibiting mitochondrial fission and GSK-3β-dependent increases in mitochondrial membrane permeability: Oncotarget, 2017; 8; 44682-93
74. Slupe AM, Merrill RA, Flippo KH, A calcineurin docking motif (LXVP) in dynamin-related protein 1 contributes to mitochondrial fragmentation and ischemic neuronal injury: J Biol Chem, 2013; 288; 12353-65
75. Wilkinson KA, Henley JM, Mechanisms, regulation and consequences of protein SUMOylation: Biochem J, 2010; 428; 133-45
76. Yeh ET, SUMOylation and De-SUMOylation: wrestling with life’s processes: J Biol Chem, 2009; 284; 8223-27
77. Guo C, Hildick KL, Luo J, SENP3-mediated deSUMOylation of dynamin-related protein 1 promotes cell death following ischaemia: EMBO J, 2013; 32; 1514-28
78. Delettre C, Griffoin JM, Kaplan J, Mutation spectrum and splicing variants in the OPA1 gene: Hum Genet, 2001; 109; 584-91
79. Song Z, Chen H, Fiket M, OPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L: J Cell Biol, 2007; 178; 749-55
80. Ehses S, Raschke I, Mancuso G, Regulation of OPA1 processing and mitochondrial fusion by m-AAA protease isoenzymes and OMA1: J Cell Biol, 2009; 187; 1023-36
81. Baburamani AA, Hurling C, Stolp H, Mitochondrial optic atrophy (OPA) 1 processing is altered in response to neonatal hypoxic-ischemic brain injury: Int J Mol Sci, 2015; 16; 22509-26
82. Zhang Z, Yu J, NR4A1 promotes cerebral ischemia reperfusion injury by repressing Mfn2-mediated mitophagy and inactivating the MAPK-ERK-CREB signaling pathway: Neurochem Res, 2018; 43; 1963-77
83. Grohm J, Kim SW, Mamrak U: Cell Death Differ, 2012; 19; 1446-58
84. Wang J, Wang P, Li S, Mdivi-1 prevents apoptosis induced by ischemia-reperfusion injury in primary hippocampal cells via inhibition of reactive oxygen species-activated mitochondrial pathway: J Stroke Cerebrovasc Dis, 2014; 23; 1491-99
85. Qi X, Qvit N, Su YC, Mochly-Rosen D, A novel Drp1 inhibitor diminishes aberrant mitochondrial fission and neurotoxicity: J Cell Sci, 2013; 126; 789-802
86. Guo X, Sesaki H, Qi X: Biochem J, 2014; 461; 137-46
87. Cho DH, Nakamura T, Fang J, S-nitrosylation of Drp1 mediates beta-amyloid-related mitochondrial fission and neuronal injury: Science, 2009; 324; 102-5
88. Flippo KH, Gnanasekaran A, Perkins GA, AKAP1 protects from cerebral ischemic stroke by inhibiting Drp1-dependent mitochondrial fission: J Neurosci, 2018; 38; 8233-42
89. Wang H, Zheng S, Liu M, The effect of propofol on mitochondrial fission during oxygen-glucose deprivation and reperfusion injury in rat hippocampal neurons: PLoS One, 2016; 11; e0165052
90. Xu P, Zhang G, Sha L, Hou S, DUSP1 alleviates cerebral ischaemia reperfusion injury via inactivating JNK-Mff pathways and repressing mitochondrial fission: Life Sci, 2018; 210; 251-62
91. Zhao H, Luo Y, Chen L, Sirt3 inhibits cerebral ischemia-reperfusion injury through normalizing Wnt/β-catenin pathway and blocking mitochondrial fission: Cell Stress Chaperones, 2018; 23; 1079-92
92. Martorell-Riera A, Segarra-Mondejar M, Muñoz JP, Mfn2 downregulation in excitotoxicity causes mitochondrial dysfunction and delayed neuronal death: EMBO J, 2014; 33; 2388-407
93. Zhang L, He Z, Zhang Q, Exercise pretreatment promotes mitochondrial dynamic protein OPA1 expression after cerebral ischemia in rats: Int J Mol Sci, 2014; 15; 4453-63
94. Tang Y, Liu X, Zhao J, Hypothermia-induced ischemic tolerance is associated with Drp1 inhibition in cerebral ischemia-reperfusion injury of mice: Brain Res, 2016; 1646; 73-83
95. Veeranki S, Tyagi SC, Mdivi-1 induced acute changes in the angiogenic profile after ischemia-reperfusion injury in female mice: Physiol Rep, 2017; 5; e13298
96. Zhang R, Xu L, Zhang D, Cardioprotection of ginkgolide B on myocardial ischemia/reperfusion-induced inflammatory injury via regulation of A20-NF-κB pathway: Front Immunol, 2018; 9; 2844
97. Zhao Q, Guo J, Cui G, Chitosan derivatives as green corrosion inhibitors for P110 steel in a carbon dioxide environment: Colloids Surf B Biointerfaces, 2020; 194; 111150
98. Pane B, Gazzola V, Spinella G, Inflammatory response modulation through a PPARγ agonist during surgically induced visceral ischemia in an animal model: Ann Vasc Surg, 2018; 48; 189-94
99. Wu X, Li X, Liu Y, Hydrogen exerts neuroprotective effects on OGD/R damaged neurons in rat hippocampal by protecting mitochondrial function via regulating mitophagy mediated by PINK1/Parkin signaling pathway: Brain Res, 2018; 1698; 89-98
100. Chen Z, Ma Y, Yang Q, AKAP1 mediates high glucose-induced mitochondrial fission through the phosphorylation of Drp1 in podocytes: J Cell Physiol, 2020; 235; 7433-48
101. Xiao F, Lv J, Liang YB, The expression of glucose transporters and mitochondrial division and fusion proteins in rats exposed to hypoxic preconditioning to attenuate propofol neurotoxicity: Int J Neurosci, 2020; 130; 161-69
102. Sheng J, Li H, Dai Q, DUSP1 recuses diabetic nephropathy via repressing JNK-Mff-mitochondrial fission pathways: J Cell Physiol, 2019; 234; 3043-57
103. Yu LM, Dong X, Xue XD, Melatonin attenuates diabetic cardiomyopathy and reduces myocardial vulnerability to ischemia-reperfusion injury by improving mitochondrial quality control: Role of SIRT6: J Pineal Res, 2020 [Online ahead of print]
104. Bei Y, Xu T, Lv D, Correction to: Exercise-induced circulating extracellular vesicles protect against cardiac ischemia-reperfusion injury: Basic Res Cardiol, 2019; 114; 44
105. Fukui Y, Nozawa T, Ihori H, Nicorandil attenuates ischemia-reperfusion injury via inhibition of norepinephrine release from cardiac sympathetic nerve terminals: Int Heart J, 2017; 58; 787-93
106. Carreira R, Monteiro P, Gonçalves LM, Providência LA, Nicorandil preserves the function of the mitochondrial phosphorylative and oxidative system in an animal model of global ischemia-reperfusion: Rev Port Cardiol, 2007; 26; 521-28
107. Paillard M, Csordás G, Huang KT: Mol Cell, 2018; 72; 778-85.e3
108. Kim MJ, Moon CH, Kim MY: Eur J Pharmacol, 2005; 525; 1-7
Figures
Figure 1. Drp1 phosphorylation/dephosphorylation and mitochondrial fission in neurons upon I/R. I/R activates Cdk1/cyclin B to induce the phosphorylation of Drp1 at serine 616, leading to mitochondrial fission via and promoting the activation/recruitment of Drp1. I/R promotes PINK1 expression to suppress the Drp1 phosphorylation at serine 616, suppressing the activation/recruitment of Drp1 and mitochondrial fission. I/R activates calcineurin, leading to Drp1 Ser637/656 dephosphorylation, increasing Drp1 activity and promoting mitochondrial fission.Figure 2. Drp1 SUMOylation/deSUMOylation and mitochondrial fission in neurons upon I/R. During ischemia, Drp1 SUMOylation by SUMO-2/3 contributes to reduced mitochondrial fission; however, after reperfusion, SENP3 levels are restored, leading to the deSUMOylation of Drp1-SUMO2/3 and increased mitochondrial fission. In Press
15 Apr 2024 : Laboratory Research
The Role of Copper-Induced M2 Macrophage Polarization in Protecting Cartilage Matrix in OsteoarthritisMed Sci Monit In Press; DOI: 10.12659/MSM.943738
07 Mar 2024 : Clinical Research
Knowledge of and Attitudes Toward Clinical Trials: A Questionnaire-Based Study of 179 Male Third- and Fourt...Med Sci Monit In Press; DOI: 10.12659/MSM.943468
08 Mar 2024 : Animal Research
Modification of Experimental Model of Necrotizing Enterocolitis (NEC) in Rat Pups by Single Exposure to Hyp...Med Sci Monit In Press; DOI: 10.12659/MSM.943443
18 Apr 2024 : Clinical Research
Comparative Analysis of Open and Closed Sphincterotomy for the Treatment of Chronic Anal Fissure: Safety an...Med Sci Monit In Press; DOI: 10.12659/MSM.944127
Most Viewed Current Articles
17 Jan 2024 : Review article
Vaccination Guidelines for Pregnant Women: Addressing COVID-19 and the Omicron VariantDOI :10.12659/MSM.942799
Med Sci Monit 2024; 30:e942799
14 Dec 2022 : Clinical Research
Prevalence and Variability of Allergen-Specific Immunoglobulin E in Patients with Elevated Tryptase LevelsDOI :10.12659/MSM.937990
Med Sci Monit 2022; 28:e937990
16 May 2023 : Clinical Research
Electrophysiological Testing for an Auditory Processing Disorder and Reading Performance in 54 School Stude...DOI :10.12659/MSM.940387
Med Sci Monit 2023; 29:e940387
01 Jan 2022 : Editorial
Editorial: Current Status of Oral Antiviral Drug Treatments for SARS-CoV-2 Infection in Non-Hospitalized Pa...DOI :10.12659/MSM.935952
Med Sci Monit 2022; 28:e935952