04 January 2022: Animal Study
Activation of α7 Nicotinic Acetylcholine Receptor by its Selective Agonist Improved Learning and Memory of Amyloid Precursor Protein/Presenilin 1 (APP/PS1) Mice via the Nrf2/HO-1 Pathway
Kun Cao12BCEF, Jie Xiang13CF, Yang-Ting Dong34BCD, Yi Xu13CD, Zhi-Zhong Guan134ADG*DOI: 10.12659/MSM.933978
Med Sci Monit 2022; 28:e933978






Abstract
BACKGROUND: To reveal the mechanism underlying the effect of α7 nicotinic acetylcholine receptor (nAChR) on neurodegeneration in Alzheimer disease (AD), the influence of the receptor on recognition in APP/PS1 mice was evaluated by using its selective agonist (PNU-282987).
MATERIAL AND METHODS: APP/PS1 and wild-type (WT) mice were treated with PNU or saline, respectively, for 7 days at the ages of 6 and 10 months.
RESULTS: Morris water maze analysis showed that both at 6 and 10 months of age, PNU treatment enhanced the learning and memory of APP/PS1 mice. However, PNU treatment did not alter the number of senile plaques. Furthermore, a higher protein expression of Nrf2/HO-1, ADAM10, SYP, and SNAP-25, and a lower level of oxidative stress, were observed in the hippocampus of APP/PS1 mice treated with PNU compared with the control group.
CONCLUSIONS: The results indicated that the activation of α7 nAChR by PNU improved the learning and memory of mice carrying the APP/PS1 mutation, regulated the levels of enzymes that mediate APP metabolization to reduce β-amyloid peptide damage, and decreased the level of oxidative stress and maintained synaptic plasticity, in which the mechanism might be enhancement of the Nrf2/HO-1 pathway.
Keywords: Alzheimer Disease, beta-Amyloid Peptide (29-42), learning disabilities, Amyloid beta-Protein Precursor, Animals, Benzamides, Bridged Bicyclo Compounds, Disease Models, Animal, Heme Oxygenase-1, Learning, Maze Learning, Membrane Proteins, Memory, Mice, Mice, Transgenic, NF-E2-Related Factor 2, Neuronal Plasticity, Nicotinic Agonists, Presenilin-1, Signal Transduction, alpha7 Nicotinic Acetylcholine Receptor
Background
Alzheimer disease (AD) is defined as significant and progressive memory loss in combination with cognitive decline and change in personality [1]. The β-amyloid peptide (Aβ) hypotheses states that AD is initiated from abnormal processing of the amyloid precursor protein (APP), causing an abundant aggregation and deposition of Aβ, and subsequently induced senile plaques formation and neuronal death [2]. However, this hypothesis has been controversial, with no drugs related to the amyloid hypothesis approved to date [3]. Meanwhile, the failures of many anti-amyloid treatments have challenged the Aβ hypothesis [4]. Senile plaques may play a protective role, such as regulating synaptic function and cellular signaling pathways, as well as possessing antioxidant properties [5]. Despite decades of research in this area and efforts by the pharmaceutical industry, there is still no effective cure for AD or even therapy to effectively retard progression of its symptoms [6,7].
Neuronal nicotinic acetylcholine receptors (nAChRs) are expressed widely throughout the human brain. These receptors have been implicated in many physiological and behavioral processes, such as cognitive enhancement and anxiety reduction, as well as neuroprotection [8]. Since cholinergic deficits are key elements of both cognitive and behavioral alterations in patients with AD, enhancement of nicotinic cholinergic transmission has been proposed as a treatment target [9]. However, the usefulness of drugs designed for this purpose may be limited because of addiction and adverse effects [10].
Among the various subtypes, α7 nAChR appears to have a key role in neurodegeneration and cognitive deficit [11]. This subtype also appears to mediate the protective effect of nicotine without being involved in its addictive properties [12]. In this context, expression of α7 nAChR in the brains of patients with neurodegenerative diseases has been found to be altered [13].
The α7 nAChR subtype is not only involved in cognitive function, but also inhibits formation of Aβ and promotes α-secretase cleavage of APP [14]. Recently, we reported that activation of α7 nAChR promoted endogenous αB-crystallin expression to inhibit Aβ aggregation via the PI3K/Akt pathway [15]. The cognitive potential of the hippocampus of rats infused with Aβ is impaired by a nAChR-dependent mechanism [16]. Furthermore, selective agonists of α7 nAChR improved the cognitive performance of rodents in various situations [17,18]. However, the underlying mechanism by which α7 nAChR affects cognition in AD is still unclear.
Recently, the effects of the Nrf2/HO-1 pathway have received increasing researcher attention in connection with the pathological process of AD [19–20]. Nrf2, a regulator of antioxidant response, has a protective effect on neuron and vascular degeneration in retinal ischemia-reperfusion injury [21]. HO-1, an antioxidant response element, plays a critical role in inflammation and iron homeostasis [22]. Experiments in vivo and in vitro showed that anthocyanins suppressed oxidative stress induced by Aβ oligomers and prevented cell apoptosis and neurodegeneration through the p-PI3K/Akt/GSK3β and Nrf2/HO-1 pathways [23]. Interestingly, the microglial α7 nAChR/Nrf2/HO-1 axis has a neuroprotective effect on cerebral ischemia by regulating inflammation and oxidative stress in microglia [24]. After primary glial cultures were treated with PNU (N-[(3R)-1-azabicyclo[2.2.2]oct-3-yl]-4-chlorobenzamide hydrochloride), a selective agonist of α7 nAChR, the mitochondrial mass and oxygen consumption increased without increasing oxidative stress, and these changes were eliminated with inhibition of Nrf2/HO-1 [25]. In the present study, transgenic animals (APP/PS1 mice) with AD were treated with PNU to determine whether the role of α7 nAChR in promoting cognitive function and attenuating oxidative stress and synaptic damage was involved in the Nrf2/HO-1 pathway.
Material and Methods
MATERIALS:
We purchased the following materials from the sources indicated: PNU-282987 (Sigma-Aldrich, USA), anti-synaptosome-associated protein 25 (SNAP-25), anti-NRF2, and anti-HO-1 antibodies (GTX113839, GTX103322, GTX 101147, Gentex, Inc., USA), anti-synaptophysin (SYP), anti-β-site amyloid precursor protein-cleaving protein 1 (BACE1) anti-BACE 2 antibodies (ab8049, ab183612 and ab270458, Abcam, Inc., USA); anti-Aβ (6E10, SIG-39340, BioLegend, Inc., USA), and anti-β-actin, anti-α disintegrin, and metalloproteinase 10 (ADAM10) antibodies (sc-376421and sc-28358, Santa Cruz, Inc., USA).
EXPERIMENTAL ANIMALS:
Double-transgenic APPswe/PS1 mice and wild-type (WT) mice of this same strain (as controls) were supplied by Shanghai Research Center for Model Organisms, China. These mice were bred at a specific pathogen-free animal laboratory center with 22–25°C in a 12-h light/12-dark cycle and given free access to food and water. Mouse tails were used for genotyping by standard polymerase chain reaction (PCR) protocols to ensure the presence of the expected genes. PCR primers were designed according to cDNA sequence (Table 1).
At ages of 6 or 10 months, WT and APP/PS1 mice received PNU-282987 (1 mg/kg) or physiological saline intraperitoneally for 2 days before and during the 5 acquisition days of the behavioral test (10 mice per group) [26]. After Morris water maze (MWM) testing, the animals were anesthetized using chloral hydrate (400 mg/kg) by intraperitoneal injection [27]. Death was confirmed by absence of heart rate, no breathing, and no reflexes. The brain tissues were collected for analysis. The study was approved by the Ethics Committee of Guizhou Medical University, China (No. 1702110).
THE MWM TEST FOR SPATIAL LEARNING AND MEMORY:
The spatial learning and memory of mice were assessed using the MWM test, as described previously [28]. In brief, a circular tank (diameter of 120 cm, height 50 cm) contained an escape platform (diameter 10 cm) hidden 1.0 cm below the surface of milky water.
During the trial, each mouse was allowed to swim freely for 60 s to find this submerged platform, being required to remain on the platform for 5 s during both the familiarization session and acquisition phase (4 trials/day on 5 consecutive days). During the retention phase (on the fifth day of testing), the platform was removed from the pool and each mouse was allowed up to 60 s to search for its former position, whereas the number of platform area crossings was measured. The movement of mice were video recorded. We recorded the time of the escape latency, the number of platform crossings, and the time spent at the original position of the platform for assessing memory.
HEMATOXYLIN AND EOSIN (HE) STAINING:
In brief, slides were dried, exposed sequentially to a series of aqueous ethanol solutions (100%, 95%, 90%, and 70%), and washed briefly; then the slides were stained with hematoxylin for 1 min and followed by eosin Y for 7 min. Next, dehydration was performed with a series of aqueous ethanol solutions (70%, 90%, 95%, and 100%) and the slides were finally cleared with xylene and mounted with Vectashield (Vector Laboratories, Inc., Burlingame, CA).
IMMUNOHISTOCHEMICAL STAINING:
Immunohistochemical staining was used to detect Aβ level, as described previously [26]. Seven-μm-thick sections were deparaffinized and hydrated. For antigen repair, 0.01 M citric buffer (pH 6.0) was used. Thereafter, sections were exposed to 6E10 primary antibody (diluted 1: 100, mouse) overnight at 4°C. Then, avidin-biotinylated enzyme complex was used as secondary antibody and a peroxidase reaction solution containing DAB was used for staining. Finally, sections were mounted using Vectashield (Vector Laboratories, CA). The number of senile plaques was counted under a microscope (original magnifications ×40, ×100, and ×200).
WESTERN BLOTTING ANALYSIS:
Total protein content was measured in the brain tissues. Thirty μg of protein in each sample was resolved with 10% SDS-PAGE, blotted onto PVDF membranes, probed overnight with primary antibody at 4°C, and incubated for 1 h at room temperature with horseradish peroxidase-conjugated secondary antibody. Finally, ECL Plus reagent was used to visualize peroxidase-coated bands.
BIOCHEMICAL ASSAY:
The activities of SOD and GSH-PX and content of MDA in brain tissues and serum were evaluated by use of the appropriate biochemical kits. The OD value was determined according to the manufacturer’s instructions.
STATISTICAL ANALYSIS:
Data are presented as means±standard deviation (SD) and are compared by analysis of variance (ANOVA), followed by the Tukey honestly significant difference test (HSD). Differences with a
Results
SPATIAL LEARNING AND MEMORY:
The escape latency time (Figure 1A) for the APP/PS1 mice at 6 and 10 months of age was significantly longer than that of WT animals, indicating the impaired ability of spatial learning. During the probe trial, APP/PS1 mice performed fewer platform crossings and spent less time at the original position of the platform (Figure 1B, 1C), demonstrating poorer memory. Treatment with PNU to APP/PS1 mice 6 or 10 months of age as compared to those without PNU exposure significantly reduced the escape latency time (Figure 1A), which was close to the level of WT mice, indicating that PNU can increase learning ability. PNU treatment of APP/PS1 mice as compared to those without PNU exposure obviously increased both the number of platform crossings and time spent at the original position of the platform in mice 6 and 10 months of age (Figure 1B, 1C), indicating that PNU can improve memory ability.
HISTOPATHOLOGICAL OBSERVATION IN THE MICE BRAINS:
The HE staining revealed no significantly histopathological changes in the CA1, CA3, and DG regions of the hippocampus of 6- and 10-month-old APP/PS1 mice, either PNU treated or without PNU, or WT mice (Figure 2A, 2B). In further studies, Nissl stain or the examination observed under an electronic microscope may be required to show pathological changes of neurons in the brains of mice with overexpression of APP.
SENILE PLAQUES IN THE BRAINS:
Immunohistochemical staining of the hippocampus of 6-month-old APP/PS1 mice revealed significantly more senile plaques (Figure 3D–3F) than that of WT animals (Figure 3A–3C). In addition, the hippocampus of APP/PS1 mice treated with PNU also showed obviously more senile plagues (Figure 3G–3I), which had no significant difference with those of APP/PS1 mice without treatment of PNU (Figure 3J). In addition, at 10 months of age, there were obviously more senile plaques in the hippocampus both in the APP/PS1 mice with or without PNU treatment (Figure 4), while no significant difference of the plaque numbers was shown between these 2 groups. There were more senile plaques in the 10-month-old APP/PS1 mice than in 6-month-old APP/PS1 mice with or without treatment of PNU (data not shown).
LEVELS OF ENZYMES THAT CLEAVE APP IN THE BRAINS:
In the brains of the 6-month-old APP/PS1 mice as compared to WT mice, the increased level of BACE1 but the decreased ADAM10 and BACE2 were found (Figure 5A–5C). Whereas, in the brains of the 6-month-old APP/PS1 mice treated with PNU, the declined BACE1, and raised ADAM10 and BACE2 were detected as compared to those of the 6-month-old APP/PS1 mice without treatment of PNU (Figure 5). Furthermore, the levels of these enzymes in the brains of the 10-month-old mice from different groups exhibited similar patterns to those of 6-month-old mice (Figure 6). In addition, the levels of BACE1 and ADAM10 were significantly correlated with the number of senile plaques in the hippocampus of APP/PS1 mice without PNU treatment (for BACE1: R=0.713 in 6-month-old mice, R=0.756 in 10-month-old mice, both P<0.05; for ADAM10: R=−0.661 in 6-month-old mice, R=−0.769 in 10-month-old mice, both P<0.05) (data not shown).
LEVELS OF NRF2/HO-1 IN THE BRAINS:
The Nrf2 level in the brains of 6-month-old APP/PS1 mice, but not 10-month-old mice, was significantly higher than in WT mice (Figure 6A, 6C). Significantly, the Nrf2 level in the brains of 6- or 10-month-old APP/PS1 mice treated with PNU was much higher than those of 6- or 10-month-old APP/PS1 mice without PNU treatment, respectively (Figure 6A, 6C). In the same way, the HO-1 level in the brains of 6- or 10-month-old APP/PS1 mice was significantly higher than in WT mice (Figure 6B, 6D). In addition, the HO-1 level in the brains of 6- or 10-month-old APP/PS1 mice treated with PNU was obviously higher than those of 6- or 10-month-old APP/PS1 mice without PNU treatment (Figure 6B, 6D).
THE LEVEL OF MDA AND ACTIVITIES OF SOD AND GSH-PX IN THE BRAINS AND SERUM:
In 6- or 10-month-old APP/PS1 mice, there were higher level of MDA and decreased activities of SOD and GSH-Px in brains and serum compared to those of WT mice (Tables 2–5). Interestingly, PNU treatment of APP/PS1 mice (6- or 10-month-old) attenuated the increased levels of oxidative stress both in brains and serum as compared to the APP/PS1 mice without PNU treatment (Tables 2–5).
LEVELS OF SYNAPTIC PROTEINS IN THE BRAIN:
Levels of SYP in the brains were significantly lower in 6- or 10-month-old APP/PS1 mice compared with WT mice (Figure 7A, 7C), which was obviously attenuated by PNU treatment. Compared to WT mice, the level of SNAP-25 was significantly lower in the brains of 10-month-old APP/PS1 mice, but the same as in 6-month-old mice (Figure 7B, 7D). Treatment with PNU in 6- or 10-month-old APP/PS1 mice obviously increased the level of SNAP-25 compared to the untreated APP/PS1 mice (Figure 7B, 7D).
Discussion
α7 nAChR has crucial regulatory functions in AD pathogenesis [17,29]. The treatment of α7 nAChR agonist in vivo showed its protective effect on neurons and improved the learning and memory ability of model animal. Therefore, the α7 nAChR agonist is an ideal drug for the treatment of AD. In AD pathology, Aβ shows a relatively high binding affinity to α7 nAChR, and the expression of these 2 proteins co-localizes in the cortical regions and the hippocampus of the brains of patients with AD [30]. The interaction between Aβ and α7 nAChR has been confirmed in various experimental models and in postmortem AD brains [31,32]. In the case of pathologically elevated Aβ concentration, this interaction between Aβ and α7 nAChR may cause AD [33]. The Aβ-α7 nAChR interaction also influences neurotransmission, synaptic plasticity, learning, and memory [34,35].
On the basis of previous in vitro findings, nicotine, an agonist of nAChRs, protects neurons against the toxicity of Aβ [36,37]. In experiments, nicotine reduced production of Aβ and improved the cognitive performance of rodent models of AD as assessed by various procedures [38,39]. However, nicotine is addictive and has many adverse effects, as well as minimal benefits, limiting its usefulness in this context. Therefore, the potential of other agonists of nAChRs in treating AD needs to be explored. PNU is known as a potent and, currently, the most specific agonist of α7 nAChR, while interacting negligibly with other nAChR subtypes [40].
In the 6- or 10-month-old APP/PS1 mice, the increased escape latency and the decreased number of platform crossings and time spent at the original position of the platform were significantly lessened by PNU treatment. These results showed that activation of α7 nAChR by PNU improves the learning and memory of APP/PS1 mice, which is consistent with our previous finding [41]. In that study, we did not find any change of the learning and memory of WT mice exposed with PNU as compared to that of WT mice without PNU treatment.
A previous study evaluated the influence of PNU on the learning and memory ability of transgenic mice 2 months of age with susceptibility to AD with or without stress [42]. In that study, no significant effect of PNU on spatial task acquisition was found in AD-susceptible transgenic mice. However, our present data suggest that PNU has an effect on recognition memory in transgenic AD mice. These opposite results may be related to the differences in the age of animals and the time of PNU administration.
Senile plaques are an extracellular spherical lesion containing aggregated Aβ caused by 2 proteases, β-and γ-secretase. Here, we found more plaques in the hippocampus of both 6- or 10-month-old APP/PS1 mice. However, the administration of PNU did not obviously decrease the number of plaques in the brains of AD animals, which indicates that PNU does not totally alter the histopathological changes, since the formation of senile plaques occurs gradually in AD with age, and PUN treatment cannot quickly destroy the plaques. Furthermore, the possible changes in Aβ fragments like 1-40/42 without ameliorating plaques induced by PNU could be important in further study.
BACE1, a protease that is responsible for initiating Aβ production, is significantly associated with the level of Aβ deposition in the brains of AD patients [43,44]. Inhibition of BACE1 is an important intervention to prevent and/or cure AD. In addition, ADAM10 plays a key role in inhibiting Aβ production and increasing αAPP level [10]. Moreover, αAPP is neuroprotective and nutritional, and elevating its metabolism can reduce the generation of toxic Aβ. Enhancing the expression and activity of α-secretase can effectively inhibit the formation of Aβ [45]. Interestingly, even though BACE2 is homologous to BACE1, it can cleave Aβ completely, thereby inhibiting the formation of Aβ [46].
Our present data indicate there were increased BACE1 and decreased ADAM10 and BACE2 in APP/PS1 mice. PNU treatment reduced the level of BACE1 and elevated ADAM10 and BACE2 in the brains of APP/PS1 mice, indicating that the activation of α7 nAChR interferes with the production of Aβ by regulating the enzyme activity in the APP metabolic process. In 10-month-old APP/PS1 mice treated with PNU, the higher level of ADAM10 was more obvious than in 6-month-old mice. Although there were significant correlations between BACE1 or ADAM10 with the number of senile plaques in the hippocampus of APP/PS1 mice without PNU treatment, the number of senile plaques in the animals was not decreased by PNU treatment, suggesting that short-term activation of α7 nAChR did not totally inhibit the production of Aβ and thus could not fully eliminate the formation of senile plaques in mouse brains in our experiment.
Oxidative stress leads to the inflammatory infiltration of neutrophils, the increased secretion of proteases, and the production of numerous oxidative intermediates [47]. These negative effects produced by free radicals in vivo are considered to be important in connection with the development of AD [48]. It has been demonstrated that Aβ can lead to neurological dysfunction by increasing the level of free radicals and decreasing the activity of antioxidant enzyme activity and the content of antioxidant compound [49]. In addition, oxidative stress damages many types of intracellular organic molecules, resulting in protein overoxidation and lipid peroxidation in AD patients [50,51].
Here, at 6 and 10 months of age, the MDA content of APP/PS1 mouse brains was higher, and the SOD and GSH-Px activities were lower than in WT animals. Importantly, treatment with PNU elevated the activities of SOD and GSH-Px and reduced the content of MDA. In addition, we found that PNU interfered with the oxidative stress level in serum of the APP/PS1 mice treated with PNU.
The activated Nrf2/HO-1 pathway is a recognized defense against internal and external oxidative stress. In the present study, we observed that the level of Nrf2/HO-1 pathway in the brains of APP/PS1 mice was increased, which could be a compensation to diminish the high level of oxidative stress induced by Aβ. Recently, the level of Nrf2 has been found to be elevated during the early stages of AD, but with the progression of this disease, the level declined significantly, further reducing resistance against oxidative stress [52]. In our present results, we also found that the level of Nrf2 in the brains of 10-month-old APP/PS1 mice did not increase as in 6-month-old APP/PS1 mice, which is supports the above data. Activation of α7 nAChR by exposure to PNU resulted in significantly higher levels of Nrf2/HO-1 in the brains of APP/PS1 mice, which may greatly reduce the level of oxidative stress in AD and ease the onset of cognitive deficit.
SYP, a structural transmembrane protein of synaptic vesicles, is important for nerve repair and establishing connections between neurons (ie, the structure and functions of synapses). A deficiency in this protein can cause cognitive dysfunction and plays a key role in neurodegenerative diseases [53]. SNAP-25, an evolutionarily conserved protein, is closely related to the growth of dendrites and axons, synaptic plasticity after central nervous system injury, and learning and memory [54]. In the present study we found lower levels of SYP and SNAP-25 in the brains of APP/PS1 mice were determined, but PNU administration elevated the levels of SYP and SNAP-25. These results indicate that activation of α7 nAChR can improve the plasticity of synaptic function. Previous studies have shown that Nrf2 deficiency affects neuronal metabolism, synaptic density, and cognitive function in aged mice [55]. Therefore, the activation of α7 nAChR by PNU may rescue the loss of synaptic proteins.
Many reports have indicated that activation of the Nrf2/HO-1 pathway can attenuate cognitive dysfunction. It has been demonstrated that Nrf2 is a promising target to alleviate oxidative damages and cognitive impairment caused by chronic cerebral hypoperfusion [56]. YVLLPSPK, a walnut-derived peptide, improved learning and memory in scopolamine-induced cognitive-impaired mice through a mechanism associated with PTEN-induced putative kinase 1-mediated mitophagy via the Nrf2/Keap1/HO-1 pathway [57]. Caffeic acid phenethyl ester 4-O-glucoside protects against oxidative stress-mediated neuronal cell apoptosis and scopolamine-induced cognitive impairment by activating Nrf2/HO-1 signaling [58]. Long-term carnosine treatment can ameliorate learning and memory disturbances in streptozotocin-diabetic rats through initiation of the NF-κB/Nrf2/HO-1 signaling cascade [59]. In our study, since the activation of α7 nAChR by PNU increased the level of Nrf2/HO-1 in the brains of APP/PS1 mice, we speculate that the improved learning and memory of APP/PS1 mice by stimulation of α7 nAChR might involve activation of the Nrf2/HO-1 pathway due to PNU exposure.
Conclusions
The activation of α7 nAChR stimulated by PNU can alleviate brain damages caused by increased production of Aβ in APP/PS1 transgenic mice via regulating enzyme activities related to APP metabolism, reducing oxidative stress level, and maintaining synaptic plasticity, and also improve the learning and memory ability of mice with highly-expressed APP. This neuroprotective mechanism may be related to enhancement of the Nrf2/HO-1 pathway.
Figures
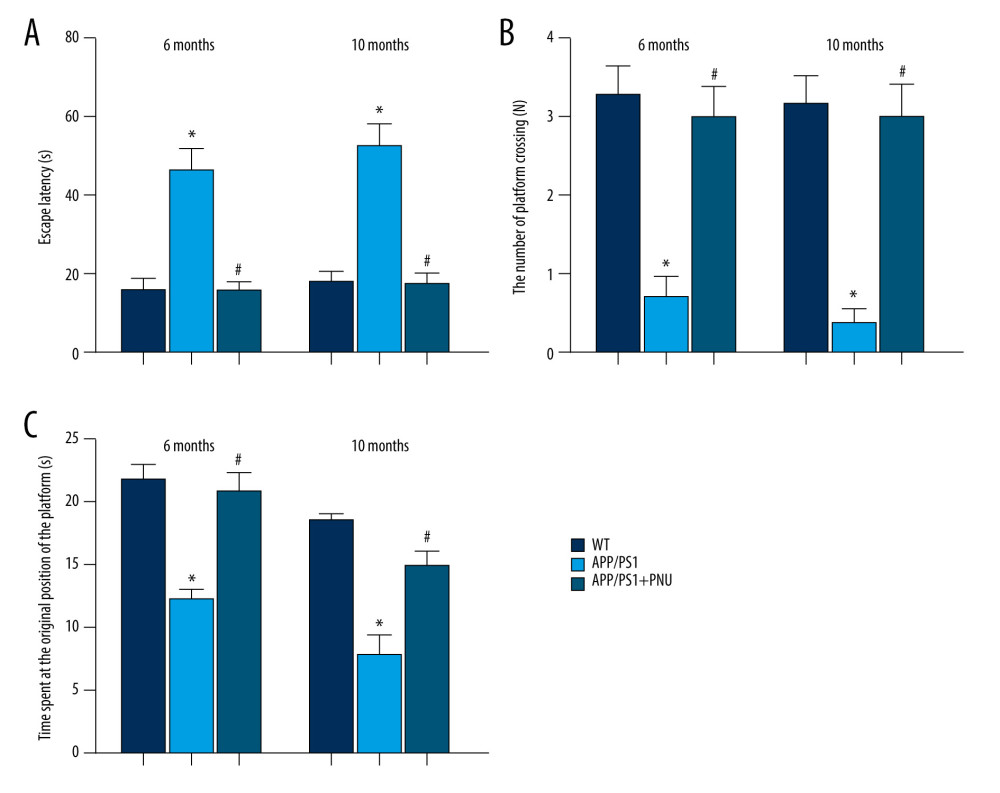 Figure 1. Effects of an activator of α7 nAChR on learning and memory in APP/PS1 mice at 6 or 10 months of age. The PNU was dissolved in sterile 0.9% saline and the pH adjusted to 7.0. The APP/PS1 mice at the ages of 6 and 10 months, respectively, were intraperitoneally received PNU (1 mg/kg) for APP/PS1 mice or physiological saline for wild-type (WT) animals for 2 days before and during the 5 acquisition days of the Morris water maze test. (A) Escape latency. (B) The number of platform crossings. (C) Time spent at the original position of platform. The values presented are means±SD of n=10 mice. * P<0.05 compared with the WT group; # P<0.05 compared with the APP/PS1 group, as determined by analysis of variance (ANOVA), followed by the Tukey HSD test.
Figure 1. Effects of an activator of α7 nAChR on learning and memory in APP/PS1 mice at 6 or 10 months of age. The PNU was dissolved in sterile 0.9% saline and the pH adjusted to 7.0. The APP/PS1 mice at the ages of 6 and 10 months, respectively, were intraperitoneally received PNU (1 mg/kg) for APP/PS1 mice or physiological saline for wild-type (WT) animals for 2 days before and during the 5 acquisition days of the Morris water maze test. (A) Escape latency. (B) The number of platform crossings. (C) Time spent at the original position of platform. The values presented are means±SD of n=10 mice. * P<0.05 compared with the WT group; # P<0.05 compared with the APP/PS1 group, as determined by analysis of variance (ANOVA), followed by the Tukey HSD test.  Figure 2. Absence of any significant histopathological changes in different regions of the hippocampus of the APP/PS1 mice at 6 or 10 months of age. (A) the APP/PS1 mice at 6 months of age. (B) the APP/PS1 mice at 10 months of age. WT – wild-type; Hippo – hippocampus; CA1/3 – Cornu Ammonis area 1/3; DG – dentate gyrus. Magnification: Hippocampus, 40×; CA1, CA2, DG regions, 200×.
Figure 2. Absence of any significant histopathological changes in different regions of the hippocampus of the APP/PS1 mice at 6 or 10 months of age. (A) the APP/PS1 mice at 6 months of age. (B) the APP/PS1 mice at 10 months of age. WT – wild-type; Hippo – hippocampus; CA1/3 – Cornu Ammonis area 1/3; DG – dentate gyrus. Magnification: Hippocampus, 40×; CA1, CA2, DG regions, 200×. 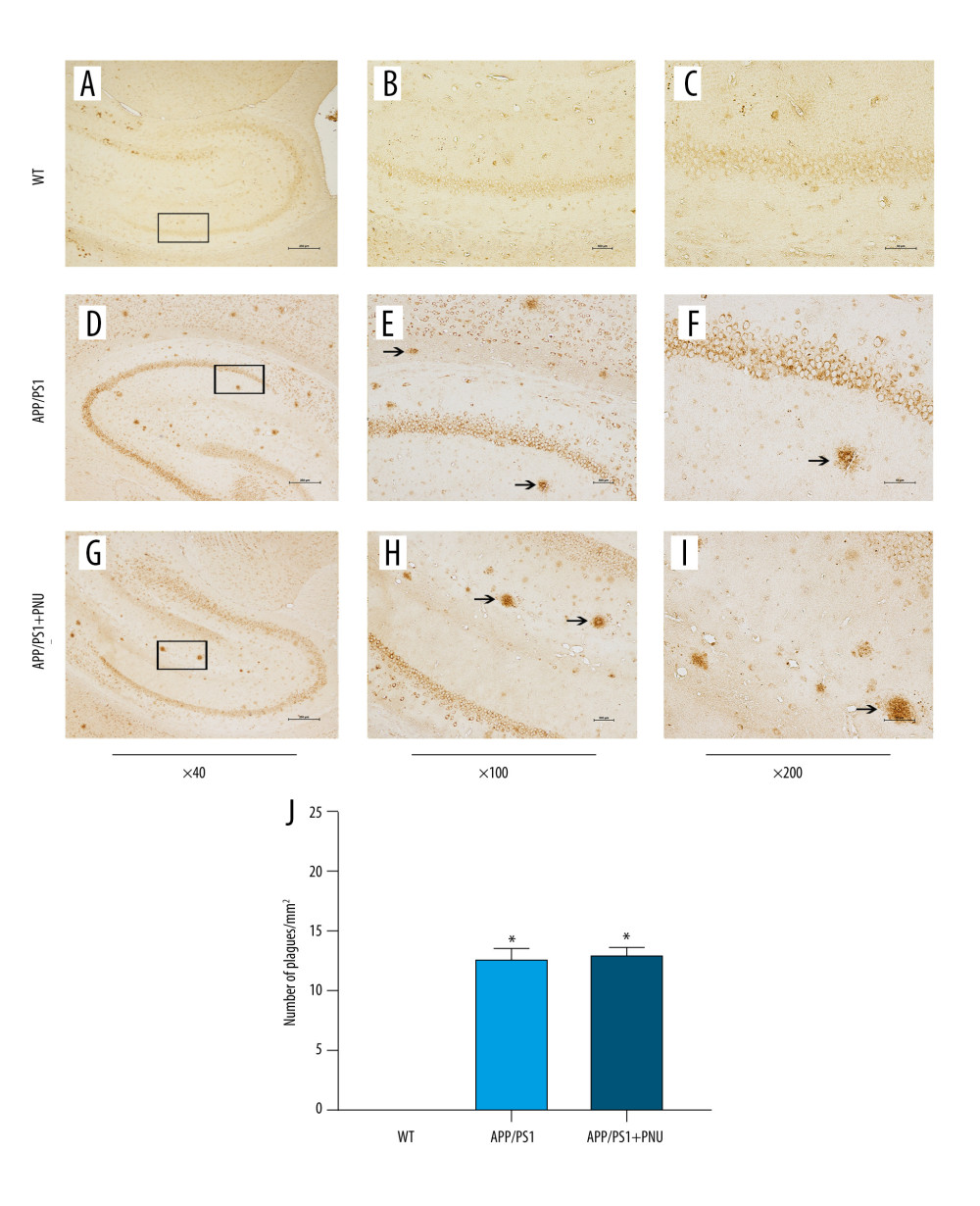 Figure 3. Immunohistochemical staining for senile plaques in the hippocampus of APP/PS1 mice at 6 months of age. (A–I) Staining of senile plaques in the hippocampus. (J) The numbers of senile plaques detected. Magnification: A, D and G, 40×, scale bar=250 μm; B, E and H, 100×, scale bar=500 μm; C, F and I, 200×, scale bar=50 μm. The solid arrows indicate senile plaques. The values presented are means±SD of n=10 mice. * P<0.05 compared to the wild-type (WT) group, as determined by analysis of variance (ANOVA), followed by the Tukey HSD test.
Figure 3. Immunohistochemical staining for senile plaques in the hippocampus of APP/PS1 mice at 6 months of age. (A–I) Staining of senile plaques in the hippocampus. (J) The numbers of senile plaques detected. Magnification: A, D and G, 40×, scale bar=250 μm; B, E and H, 100×, scale bar=500 μm; C, F and I, 200×, scale bar=50 μm. The solid arrows indicate senile plaques. The values presented are means±SD of n=10 mice. * P<0.05 compared to the wild-type (WT) group, as determined by analysis of variance (ANOVA), followed by the Tukey HSD test. 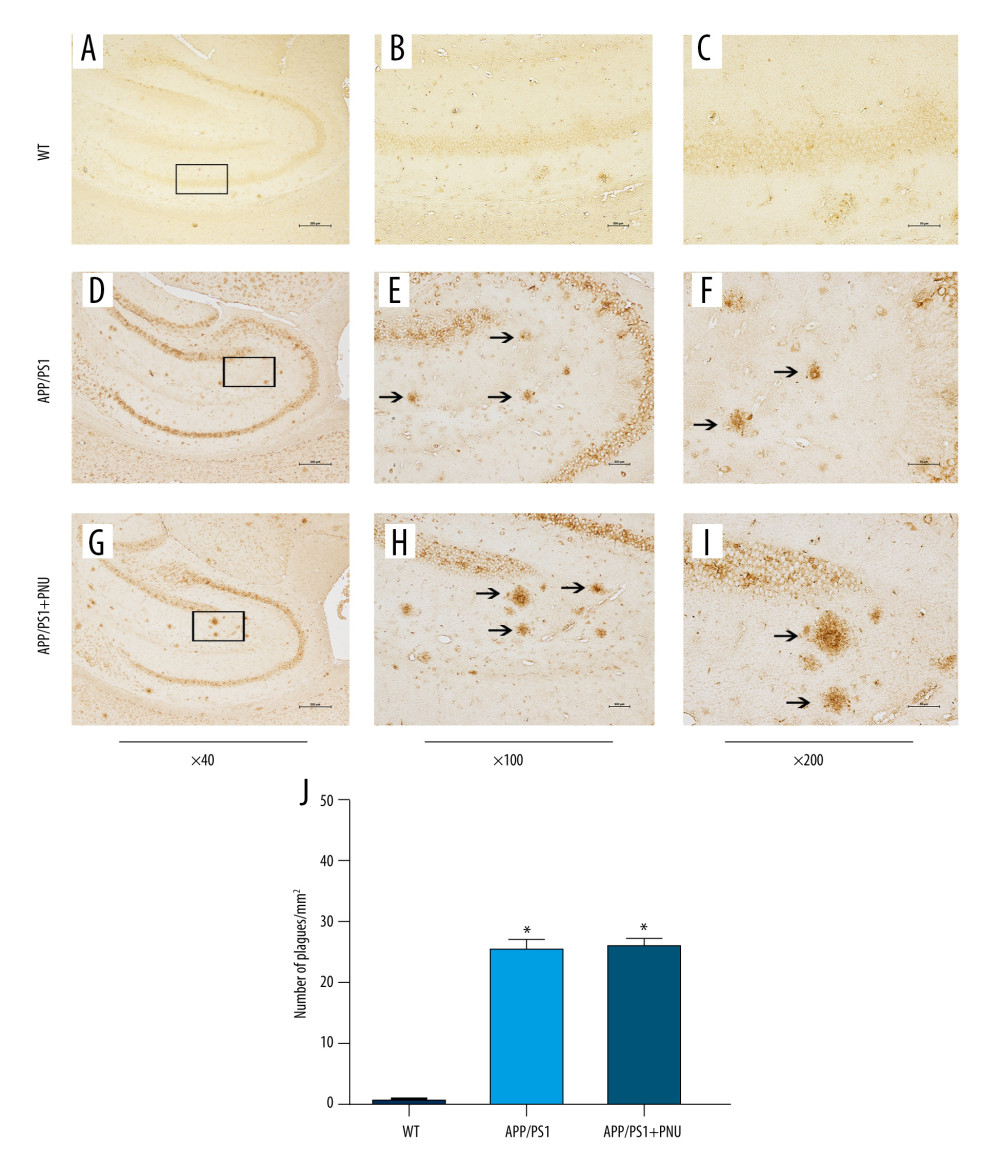 Figure 4. Immunohistochemical staining for senile plaques in the hippocampus of APP/PS1 mice at 10 months of age. (A–I) Staining senile plaques in the hippocampus. (J) The numbers of senile plaques detected. Magnification: A, D and G, 40×, scale bar=250 μm; B, E and H, 100×, scale bar=500 μm; C, F and I, 200×, scale bar=50 μm. The solid arrows indicate senile plaques. The values presented are means±SD of n=10 mice. * P<0.05 compared to the wild-type (WT) group, as determined by analysis of variance (ANOVA), followed by the Tukey HSD test.
Figure 4. Immunohistochemical staining for senile plaques in the hippocampus of APP/PS1 mice at 10 months of age. (A–I) Staining senile plaques in the hippocampus. (J) The numbers of senile plaques detected. Magnification: A, D and G, 40×, scale bar=250 μm; B, E and H, 100×, scale bar=500 μm; C, F and I, 200×, scale bar=50 μm. The solid arrows indicate senile plaques. The values presented are means±SD of n=10 mice. * P<0.05 compared to the wild-type (WT) group, as determined by analysis of variance (ANOVA), followed by the Tukey HSD test. 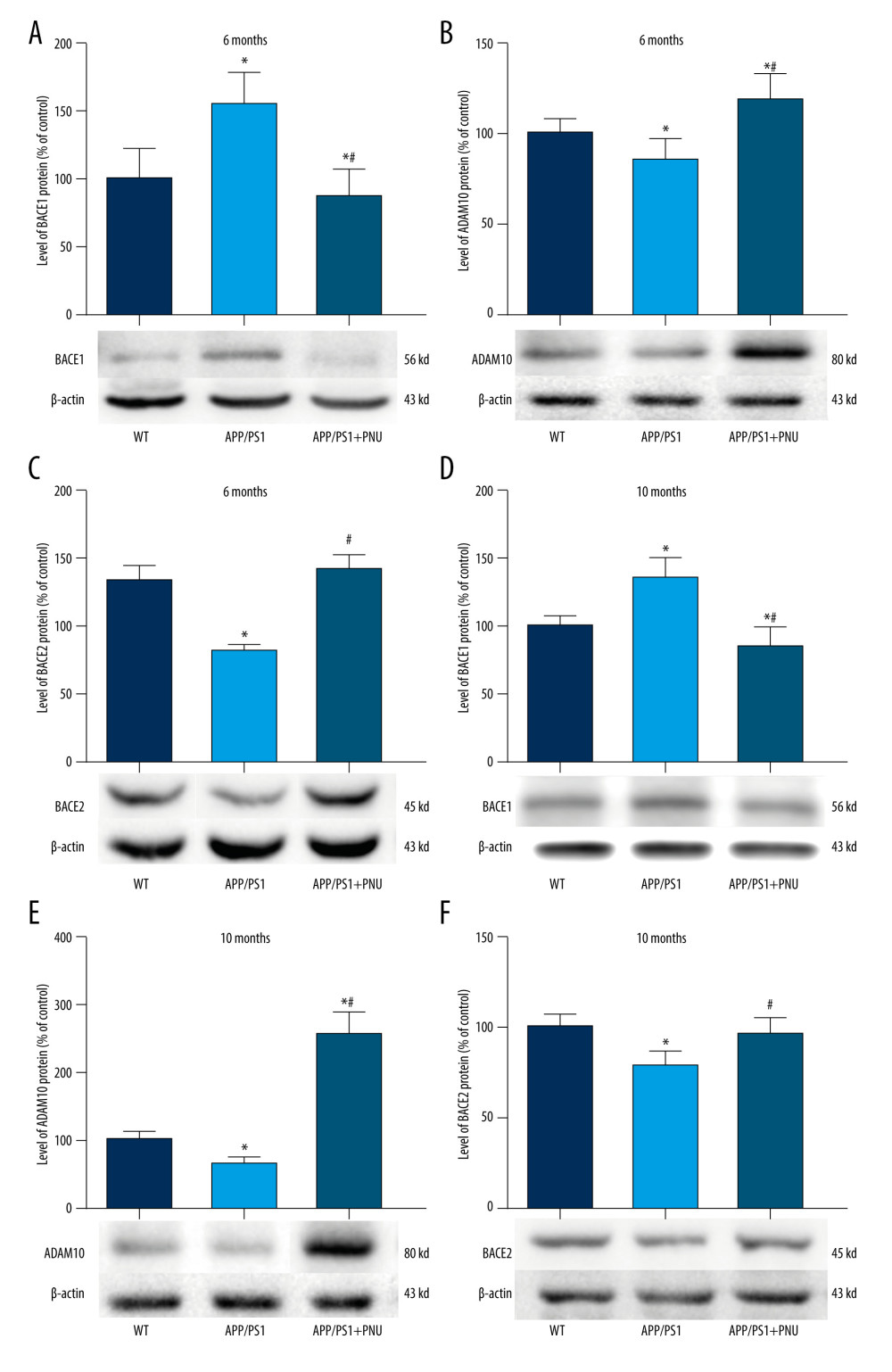 Figure 5. Western blot for BACE1, ADAM10, and BACE2 in the brains of APP/PS1 mice. (A, D) Levels of BACE1 at 6 and 10 months of age. (B, E) Levels of ADAM10 at 6 and 10 months of age. (C, F) Levels of BACE2 at 6 and 10 months of age. The values presented are means±SD of n=10 mice. * P<0.05 compared to the wild-type (WT) group; # P<0.05 compared to the APP/PS1 group, as determined by analysis of variance (ANOVA), followed by the Tukey HSD test.
Figure 5. Western blot for BACE1, ADAM10, and BACE2 in the brains of APP/PS1 mice. (A, D) Levels of BACE1 at 6 and 10 months of age. (B, E) Levels of ADAM10 at 6 and 10 months of age. (C, F) Levels of BACE2 at 6 and 10 months of age. The values presented are means±SD of n=10 mice. * P<0.05 compared to the wild-type (WT) group; # P<0.05 compared to the APP/PS1 group, as determined by analysis of variance (ANOVA), followed by the Tukey HSD test. 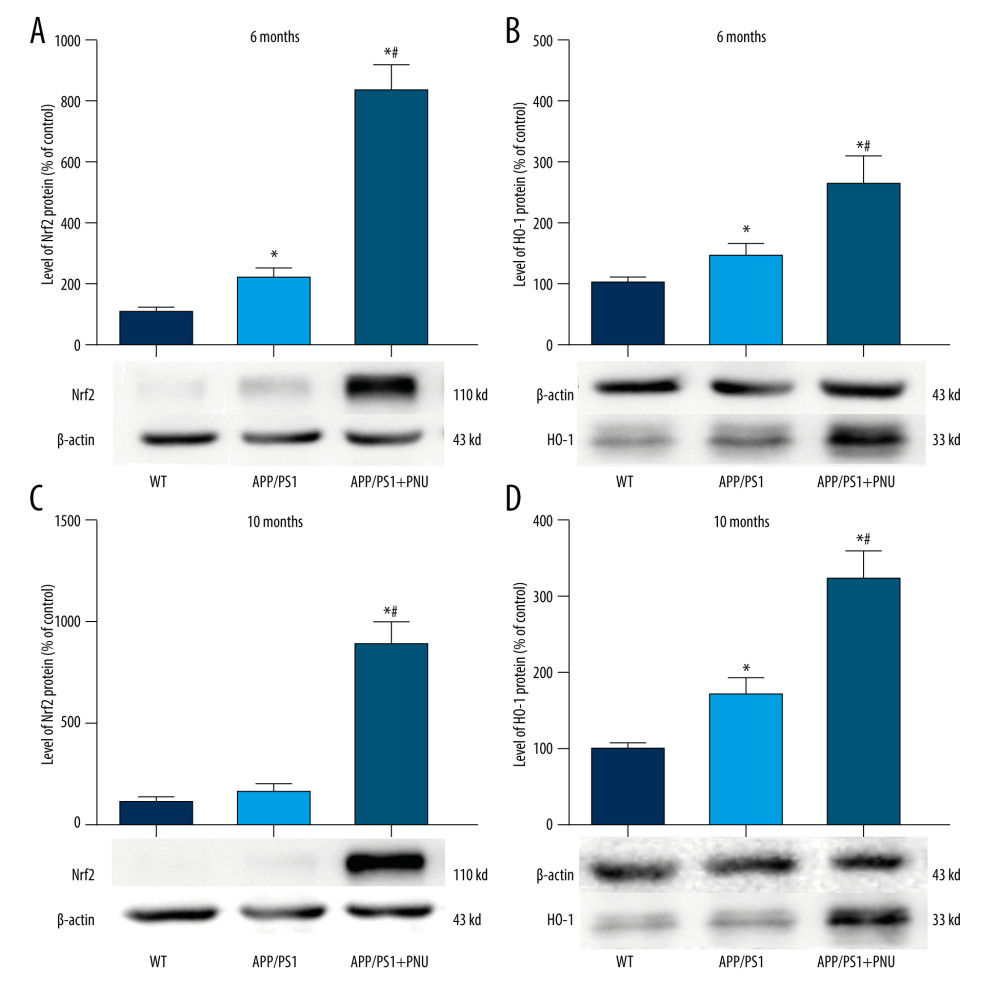 Figure 6. Western blot for Nrf2/HO-1 in the brains of APP/PS1 mice. (A, C) Nrf2 expression at 6 and 10 months of age. (B, D) HO-1 expression at 6 and 10 months of age. The values presented are means±SD of n=10 mice. * P<0.05 compared to the wild-type (WT) group; # P<0.05 compared to the APP/PS1 group, as determined by analysis of variance (ANOVA), followed by the Tukey HSD test.
Figure 6. Western blot for Nrf2/HO-1 in the brains of APP/PS1 mice. (A, C) Nrf2 expression at 6 and 10 months of age. (B, D) HO-1 expression at 6 and 10 months of age. The values presented are means±SD of n=10 mice. * P<0.05 compared to the wild-type (WT) group; # P<0.05 compared to the APP/PS1 group, as determined by analysis of variance (ANOVA), followed by the Tukey HSD test. 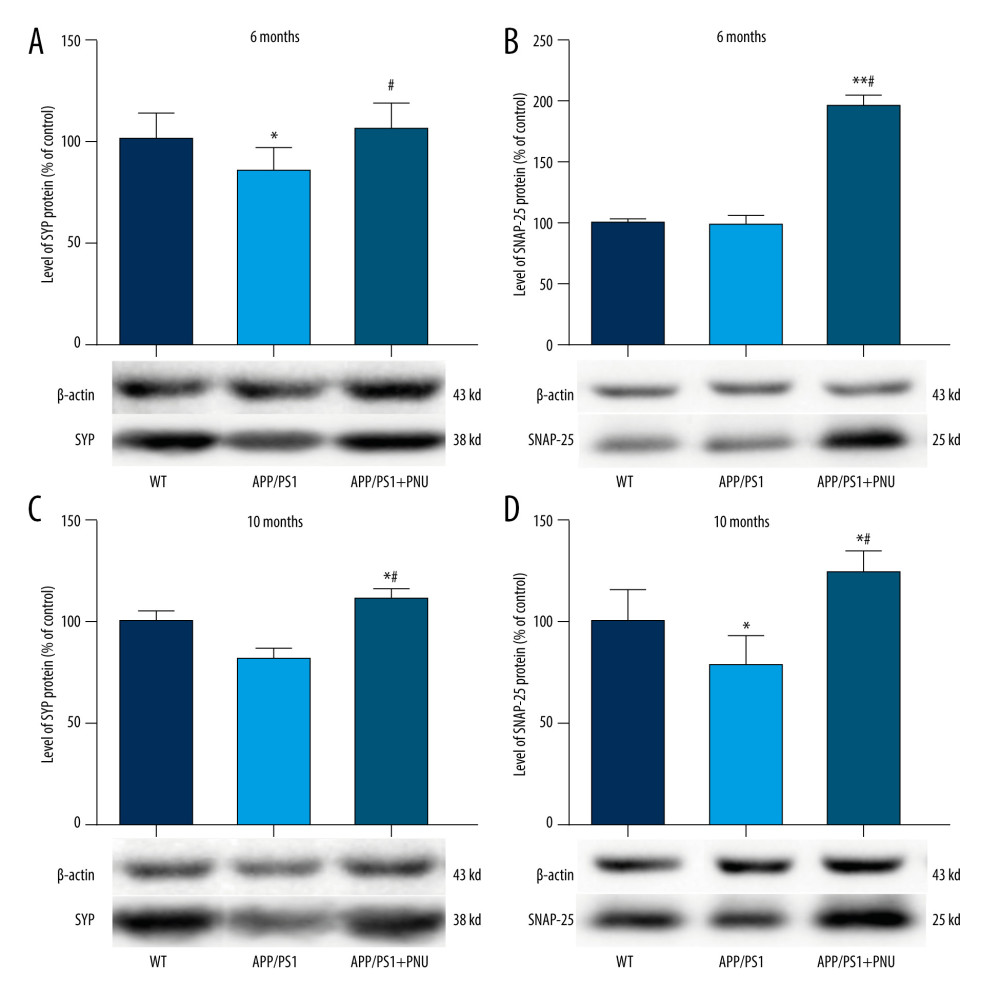 Figure 7. Western blot for SYP and SNAP-25 in the brains of APP/PS1 mice. (A, C) SYP expression at 6 and 10 months of age. (B, D) SNAP-25 expression at 6 and 10 months of age. The values presented are means±SD of n=10 mice. * P<0.05 compared to the wild-type (WT) group; # P<0.05 compared to the APP/PS1 group, as determined by analysis of variance (ANOVA), followed by the Tukey HSD test.
Figure 7. Western blot for SYP and SNAP-25 in the brains of APP/PS1 mice. (A, C) SYP expression at 6 and 10 months of age. (B, D) SNAP-25 expression at 6 and 10 months of age. The values presented are means±SD of n=10 mice. * P<0.05 compared to the wild-type (WT) group; # P<0.05 compared to the APP/PS1 group, as determined by analysis of variance (ANOVA), followed by the Tukey HSD test. Tables
Table 1. Sequences of the primers used to detect the APP and PS1 genes.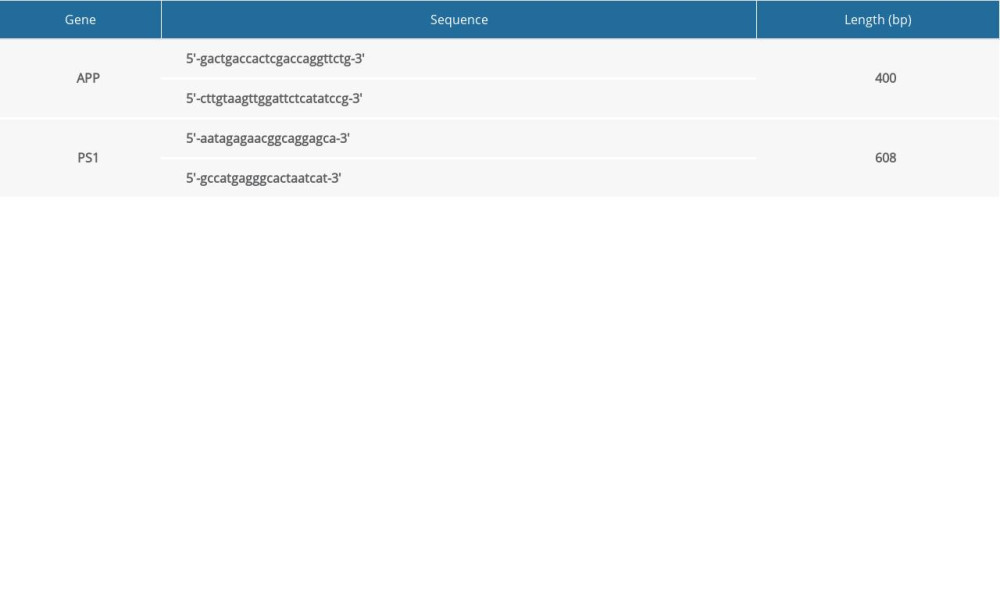 Table 2. The content of MDA and activities of SOD and GSH-Px in the brains of mice at 6 months of age.
Table 2. The content of MDA and activities of SOD and GSH-Px in the brains of mice at 6 months of age.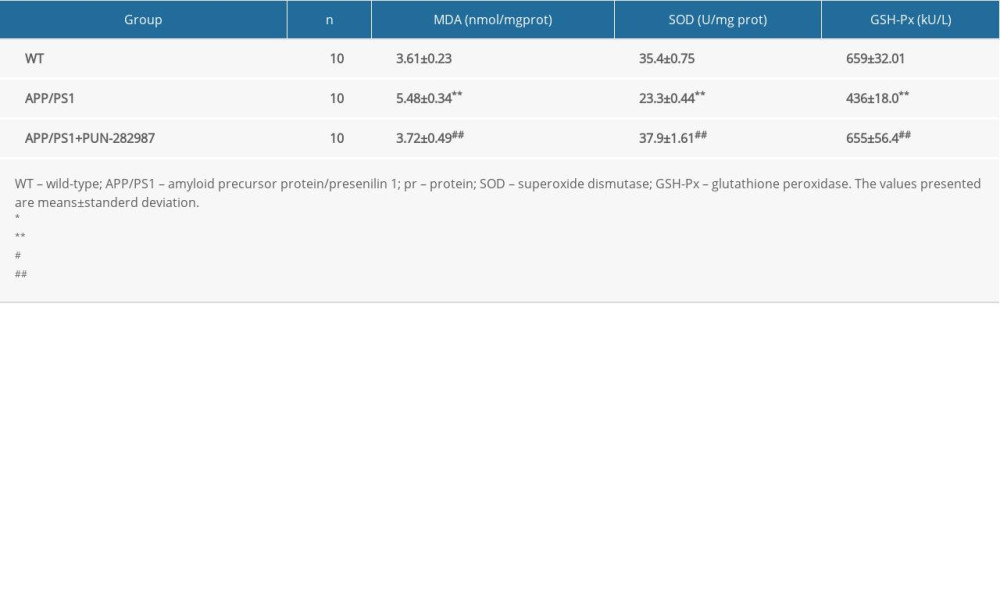 Table 3. The content of MDA and activities of SOD and GSH-Px in the brains of mice at 10 months of age.
Table 3. The content of MDA and activities of SOD and GSH-Px in the brains of mice at 10 months of age.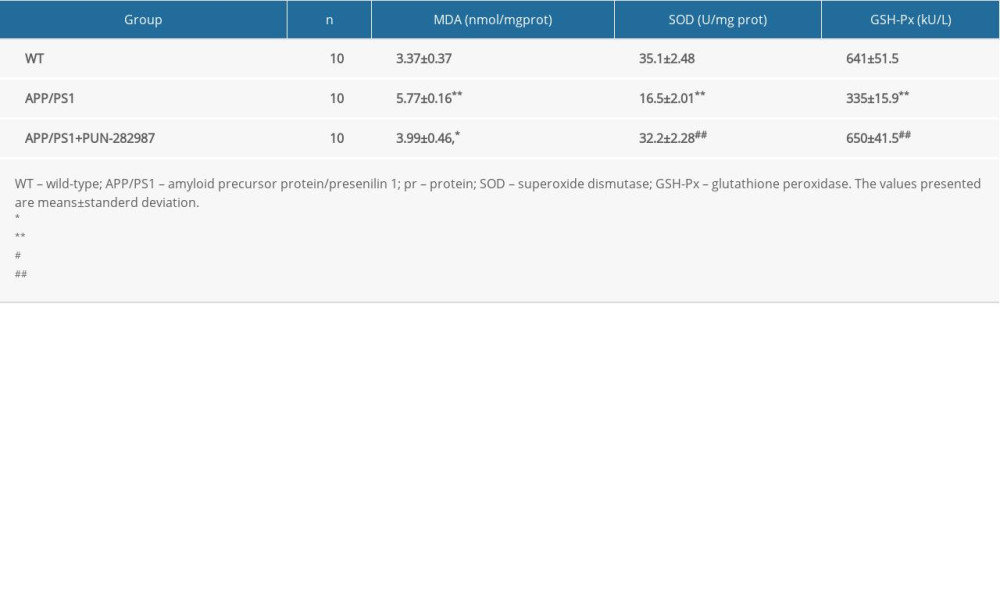 Table 4. The content of MDA and activities of SOD and GSH-Px in the serum of mice at 6 months of age.
Table 4. The content of MDA and activities of SOD and GSH-Px in the serum of mice at 6 months of age.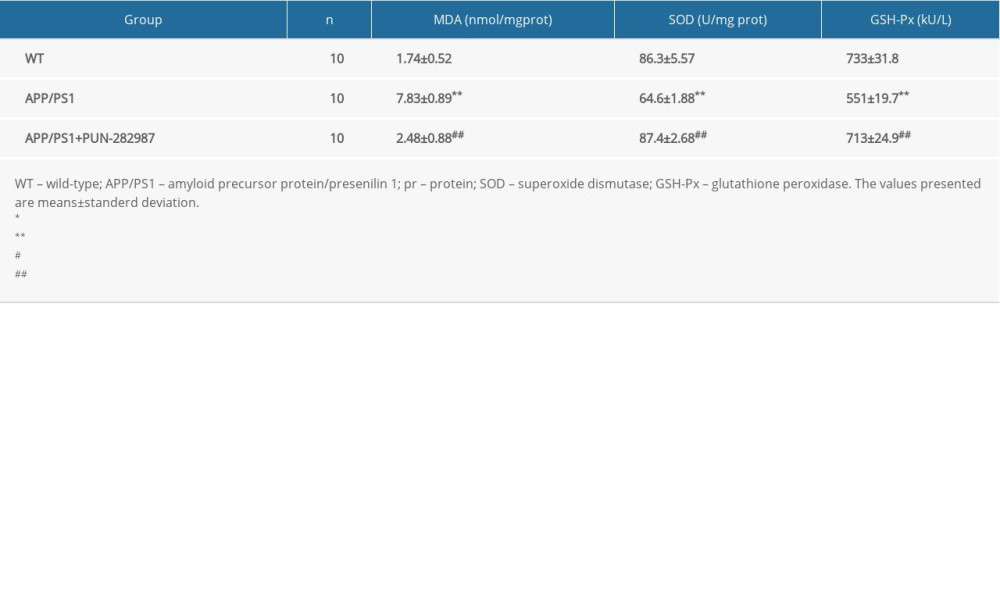 Table 5. The content of MDA and activities of SOD and GSH-Px in the serum of mice at 10 months of age.
Table 5. The content of MDA and activities of SOD and GSH-Px in the serum of mice at 10 months of age.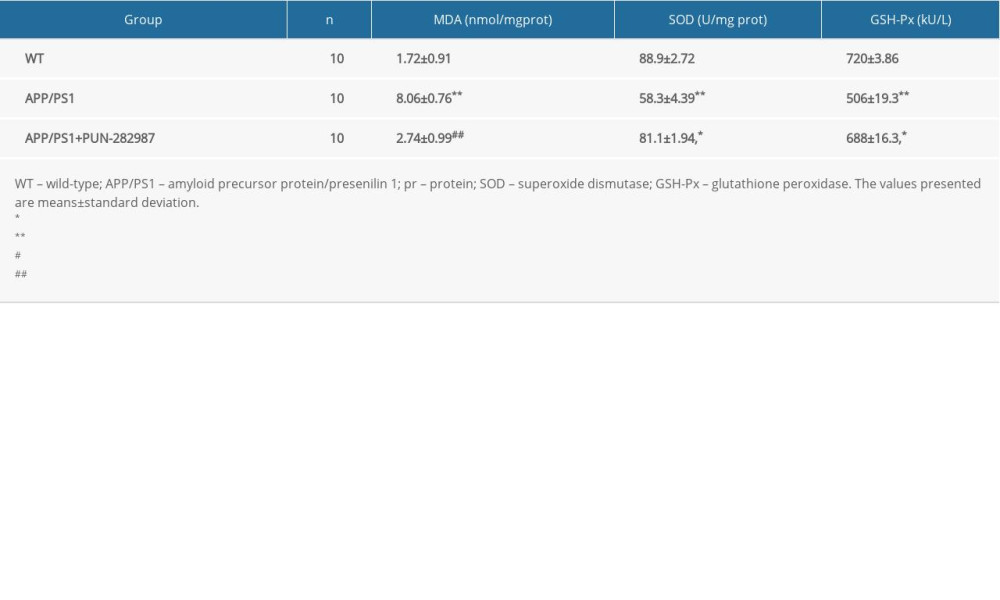
References
1. Breijyeh Z, Karaman R, Comprehensive review on Alzheimer’s disease: Causes and treatment: Molecules, 2020; 25; 5789
2. Ashrafian H, Zadeh EH, Khan RH, Review on Alzheimer’s disease: Inhibition of amyloid beta and tau tangle formation: Int J Biol Macromol, 2021; 167; 382-94
3. Imbimbo BP, Ippati S, Watling M, Should drug discovery scientists still embrace the amyloid hypothesis for Alzheimer’s disease or should they be looking elsewhere?: Expert Opin Drug Discov, 2020; 15; 1241-51
4. Tolar M, Abushakra S, Sabbagh M, The path forward in Alzheimer’s disease therapeutics: Reevaluating the amyloid cascade hypothesis: Alzheimers Dement, 2020; 16; 1553-60
5. Karisetty BC, Bhatnagar A, Armour EM, Amyloid-β peptide impact on synaptic function and neuroepigenetic gene control reveal new therapeutic strategies for Alzheimer’s disease: Front Mol Neurosci, 2020; 13; 577622
6. Vaz M, Silvestre S, Alzheimer’s disease: Recent treatment strategies: Eur J Pharmacol, 2020; 887; 173554
7. Shi J, Sabbagh MN, Vellas B, Alzheimer’s disease beyond amyloid: Strategies for future therapeutic interventions: BMJ, 2020; 371; m3684
8. Zoli M, Pucci S, Vilella A, Neuronal and extraneuronal nicotinic acetylcholine receptors: Curr Neuropharmacol, 2018; 16; 338-49
9. Akhoon BA, Choudhary S, Tiwari H, Discovery of a new donepezil-like acetylcholinesterase inhibitor for targeting Alzheimer’s disease: Computational studies with biological validation: J Chem Inf Model, 2020; 60; 4717-29
10. Verma S, Kumar A, Tripathi T, Muscarinic and nicotinic acetylcholine receptor agonists: Current scenario in Alzheimer’s disease therapy: J Pharm Pharmacol, 2018; 70; 985-93
11. Hoskin JL, Al-Hasan Y, Sabbagh MN, Nicotinic acetylcholine receptor agonists for the treatment of Alzheimer’s sementia: An update: Nicotine Tob Res, 2019; 21; 370-76
12. Farhat SM, Ahmed T, Neuroprotective and neurotoxic implications of α7 nicotinic acetylcholine receptor and Aβ interaction: therapeutic options in Alzheimer’s disease: Curr Drug Targets, 2017; 18; 1537-44
13. Yu WF, Guan ZZ, Bogdanovic N, Nordberg A, High selective expression of alpha7 nicotinic receptors on astrocytes in the brains of patients with sporadic Alzheimer’s disease and patients carrying Swedish APP 670/671 mutation: A possible association with neuritic plaques: Exp Neurol, 2005; 192; 215-25
14. Shen J, Wu J, Nicotinic cholinergic mechanisms in Alzheimer’s disease: Int Rev Neurobiol, 2015; 124; 275-92
15. Ren Z, Yang M, Guan Z, Astrocytic α7 nicotinic receptor activation inhibits amyloid-β aggregation by upregulating endogenous αB-crystallin through the PIK/AKT signaling pathway: Curr Alzheimer Res, 2019; 16; 39-48
16. Rojasgutierrez E, Muñoz-Arenas G, Treviño S, Alzheimer’s disease and metabolic syndrome: A link from oxidative stress and inflammation to neurodegeneration: Synapse, 2017; 71; e21990
17. Ma KG, Qian YH, Alpha 7 nicotinic acetylcholine receptor and its effects on Alzheimer’s disease: Neuropeptides, 2019; 73; 96-106
18. Fan H, Gu R, Wei D, The α7 nAChR selective agonists as drug candidates for Alzheimer’s disease: Adv Exp Med Biol, 2015; 827; 353-65
19. Kong D, Yan Y, He XY, Effects of resveratrol on the mechanisms of antioxidants and estrogen in Alzheimer’s disease: Biomed Res Int, 2019; 2019; 8983752
20. Morroni F, Sita G, Graziosi A, Neuroprotective effect of caffeic acid phenethyl ester in a mouse model of Alzheimer’s disease involves Nrf2/HO-1 Pathway: Aging Dis, 2018; 9; 605-22
21. Song X, Long D, Nrf2 and ferroptosis: A new research direction for neurodegenerative diseases: Front Neurosci, 2020; 14; 267
22. Facchinetti MM, Heme-oxygenase-1: Antioxid Redox Signal, 2020; 32; 1239-42
23. Ali T, Kim T, Rehman SU, Natural dietary supplementation of anthocyanins via PI3K/Akt/Nrf2/HO-1 pathways mitigate oxidative stress, neurodegeneration, and memory impairment in a mouse model of Alzheimer’s disease: Mol Neurobiol, 2018; 55; 6076-93
24. Parada E, Egea J, Buendia I, The microglial α7-acetylcholine nicotinic receptor is a key element in promoting neuroprotection by inducing heme oxygenase-1 via nuclear factor erythroid-2-related factor 2: Antioxid Redox Signal, 2013; 19; 1135-48
25. Navarro E, Gonzalez-Lafuente L, Pérez-Liébana I, Heme-oxygenase I and PCG-1α regulate mitochondrial biogenesis via microglial activation of alpha7 nicotinic acetylcholine receptors using PNU282987: Antioxid Redox Signal, 2017; 27; 93-105
26. Vicens P, Ribes D, Torrente M, Behavioral effects of PNU-282987, an alpha7 nicotinic receptor agonist, in mice: Behav Brain Res, 2011; 216; 341-48
27. Sargin D, Mercaldo V, Yiu AP, CREB regulates spine density of lateral amygdala neurons: Implications for memory allocation: Front Behav Neurosci, 2013; 7; 209
28. Dong YT, Cao K, Xiang J, SIRT1 attenuates the neurotoxicity associated with Alzheimer’s disease, via a mechanism which may involve regulation of peroxisome proliferator-activated receptor γ coactivator 1-α: Am J Pathol, 2020; 190; 1545-64
29. Cieslikiewicz-Bouet M, Naldi M, Bartolini M, Functional characterization of multifunctional ligands targeting acetylcholinesterase and alpha 7 nicotinic acetylcholine receptor: Biochem Pharmacol, 2020; 177; 114010
30. Li SF, Wu MN, Wang XH, Requirement of α7 nicotinic acetylcholine receptors for amyloid β protein-induced depression of hippocampal long-term potentiation in CA1 region of rats in vivo: Synapse, 2011; 65; 1136
31. Ren JM, Zhang SL, Wang XL, Expression levels of the α7 nicotinic acetylcholine receptor in the brains of patients with Alzheimer’s disease and their effect on synaptic proteins in SH-SY5Y cells: Mol Med Rep, 2020; 22; 2063-75
32. Li S, Selkoe DJ, A mechanistic hypothesis for the impairment of synaptic plasticity by soluble Aβ oligomers from Alzheimer’s brain: J Neurochem, 2020; 154; 583-97
33. Dineley KT, Beta-amyloid peptide-nicotinic acetylcholine receptor interaction: The two faces of health and disease: Front Biosci, 2006; 12; 5030-38
34. Baranowska U, Wiśniewska RJ, The α7-nACh nicotinic receptor and its role in memory and selected diseases of the central nervous system: Postepy Hig Med Dosw (Online), 2017; 71; 633-48
35. Hahm ET, Nagaraja RY, Waro G, Cholinergic homeostatic synaptic plasticity drives the progression of Aβ-induced changes in neural activity: Cell Rep, 2018; 24; 342-54
36. Lu X, Zhang Y, Li H: Neurosci Lett, 2021; 761; 136114
37. Guo CN, Sun L, Liu GL, Protective effect of nicotine on the cultured rat basal forebrain neurons damaged by β-Amyloid (Aβ)25–35 protein cytotoxicity: Eur Rev Med Pharmacol Sci, 2015; 19; 2964-72
38. Majdi A, Kamari F, Vafaee MS, Revisiting nicotine’s role in the ageing brain and cognitive impairment: Rev Neurosci, 2017; 28; 767-81
39. Boiangiu RS, Mihasan M, Gorgan DL, Cotinine and 6-hydroxy-L-nicotine reverses memory deficits and reduces oxidative stress in Aβ25–35-induced rat model of Alzheimer’s disease: Antioxidants (Basel), 2020; 9; 768
40. Chan WK, Wong PT, Sheu FS, Frontal cortical alpha7 and alpha4beta2 nicotinic acetylcholine receptors in working and reference memory: Neuropharmacology, 2007; 52; 1641-49
41. Wang XL, Deng YX, Gao YM, Activation of α7 nAChR by PNU-282987 improves synaptic and cognitive functions through restoring the expression of synaptic-associated proteins and the CaM-CaMKII-CREB signaling pathway: Aging (Albany NY), 2020; 12; 543-70
42. Vicens P, Ribes D, Heredia L, Effects of an alpha7 nicotinic receptor agonist and stress on spatial memory in an animal model of Alzheimer’s disease: Biomed Res Int, 2013; 952719
43. Hampel H, Vassar R, De Strooper B, The β-secretase BACE1 in Alzheimer’s disease: Biol Psychiatry, 2021; 89; 745-56
44. Imbimbo BP, Ippati S, Watling M, Should drug discovery scientists still embrace the amyloid hypothesis for Alzheimer’s disease or should they be looking elsewhere?: Expert Opin Drug Discov, 2020; 15; 1241-51
45. Panza F, Lozupone M, Logroscino G, A critical appraisal of amyloid-β-targeting therapies for Alzheimer disease: Nat Rev Neurol, 2019; 15; 73-88
46. Marttinen M, Kurkinen KM, Soininen H, Synaptic dysfunction and septin protein family members in neurodegenerative diseases: Mol Neurodegener, 2015; 10; 16
47. Cheignon C, Tomas M, Bonnefont-Rousselot D, Oxidative stress and the amyloid beta peptide in Alzheimer’s disease: Redox Biol, 2018; 14; 450-64
48. Caruso G, Spampinato SF, Cardaci V, β-amyloid and oxidative stress: Perspectives in drug development: Curr Pharm Des, 2019; 25; 4771-81
49. Kumar A, Singh A, Ekavali , A review on Alzheimer’s disease pathophysiology and its management: An update: Pharmacol Rep, 2015; 67; 195-203
50. Singh A, Kukreti R, Saso L, Oxidative stress: A key modulator in neurodegenerative diseases: Molecules, 2019; 24(8); 1583
51. Butterfield DA, Halliwell B, Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease: Nat Rev Neurosci, 2019; 20; 148-60
52. Komaravelli N, Tian B, Ivanciuc T, Respiratory syncytial virus infection down-regulates antioxidant enzyme expression by triggering deacetylation-proteasomal degradation of Nrf2: Free Radic Biol Med, 2014; 88(Pt B); 391-403
53. Longhena F, Faustini G, Brembati V, An updated reappraisal of synapsins: Structure, function and role in neurological and psychiatric disorders: Neurosci Biobehav Rev, 2021; 130; 33-60
54. Kádková A, Radecke J, Sørensen JB, The SNAP-25 protein family: Neuroscience, 2019; 420; 50-71
55. Zweig JA, Caruso M, Brandes MS, Loss of NRF2 leads to impaired mitochondrial function, decreased synaptic density and exacerbated age-related cognitive deficits: Exp Gerontol, 2020; 131; 110767
56. Yu A, Mao L, Zhao F, Olfactory ensheathing cells transplantation attenuates chronic cerebral hypoperfusion induced cognitive dysfunction and brain damages by activating Nrf2/HO-1 signaling pathway: Am J Transl Res, 2018; 10; 3111-21
57. Zhao F, Liu C, Fang L, Walnut-derived peptide activates PINK1 via the NRF2/KEAP1/HO-1 pathway, promotes mitophagy, and alleviates learning and memory impairments in a mice model: J Agric Food Chem, 2021; 69; 2758-72
58. Wan T, Wang Z, Luo Y, FA-97, a new synthetic caffeic acid phenethyl ester derivative, protects against oxidative stress-mediated neuronal cell apoptosis and scopolamine-induced cognitive impairment by activating Nrf2/HO-1 signaling: Oxid Med Cell Longev, 2019; 2019; 8239642
59. Ahshin-Majd S, Zamani S, Kiamari T, Carnosine ameliorates cognitive deficits in streptozotocin-induced diabetic rats: Possible involved mechanisms: Peptides, 2016; 86; 102-11
Figures
Figure 1. Effects of an activator of α7 nAChR on learning and memory in APP/PS1 mice at 6 or 10 months of age. The PNU was dissolved in sterile 0.9% saline and the pH adjusted to 7.0. The APP/PS1 mice at the ages of 6 and 10 months, respectively, were intraperitoneally received PNU (1 mg/kg) for APP/PS1 mice or physiological saline for wild-type (WT) animals for 2 days before and during the 5 acquisition days of the Morris water maze test. (A) Escape latency. (B) The number of platform crossings. (C) Time spent at the original position of platform. The values presented are means±SD of n=10 mice. * P<0.05 compared with the WT group; # P<0.05 compared with the APP/PS1 group, as determined by analysis of variance (ANOVA), followed by the Tukey HSD test.Figure 2. Absence of any significant histopathological changes in different regions of the hippocampus of the APP/PS1 mice at 6 or 10 months of age. (A) the APP/PS1 mice at 6 months of age. (B) the APP/PS1 mice at 10 months of age. WT – wild-type; Hippo – hippocampus; CA1/3 – Cornu Ammonis area 1/3; DG – dentate gyrus. Magnification: Hippocampus, 40×; CA1, CA2, DG regions, 200×.Figure 3. Immunohistochemical staining for senile plaques in the hippocampus of APP/PS1 mice at 6 months of age. (A–I) Staining of senile plaques in the hippocampus. (J) The numbers of senile plaques detected. Magnification: A, D and G, 40×, scale bar=250 μm; B, E and H, 100×, scale bar=500 μm; C, F and I, 200×, scale bar=50 μm. The solid arrows indicate senile plaques. The values presented are means±SD of n=10 mice. * P<0.05 compared to the wild-type (WT) group, as determined by analysis of variance (ANOVA), followed by the Tukey HSD test.Figure 4. Immunohistochemical staining for senile plaques in the hippocampus of APP/PS1 mice at 10 months of age. (A–I) Staining senile plaques in the hippocampus. (J) The numbers of senile plaques detected. Magnification: A, D and G, 40×, scale bar=250 μm; B, E and H, 100×, scale bar=500 μm; C, F and I, 200×, scale bar=50 μm. The solid arrows indicate senile plaques. The values presented are means±SD of n=10 mice. * P<0.05 compared to the wild-type (WT) group, as determined by analysis of variance (ANOVA), followed by the Tukey HSD test.Figure 5. Western blot for BACE1, ADAM10, and BACE2 in the brains of APP/PS1 mice. (A, D) Levels of BACE1 at 6 and 10 months of age. (B, E) Levels of ADAM10 at 6 and 10 months of age. (C, F) Levels of BACE2 at 6 and 10 months of age. The values presented are means±SD of n=10 mice. * P<0.05 compared to the wild-type (WT) group; # P<0.05 compared to the APP/PS1 group, as determined by analysis of variance (ANOVA), followed by the Tukey HSD test.Figure 6. Western blot for Nrf2/HO-1 in the brains of APP/PS1 mice. (A, C) Nrf2 expression at 6 and 10 months of age. (B, D) HO-1 expression at 6 and 10 months of age. The values presented are means±SD of n=10 mice. * P<0.05 compared to the wild-type (WT) group; # P<0.05 compared to the APP/PS1 group, as determined by analysis of variance (ANOVA), followed by the Tukey HSD test.Figure 7. Western blot for SYP and SNAP-25 in the brains of APP/PS1 mice. (A, C) SYP expression at 6 and 10 months of age. (B, D) SNAP-25 expression at 6 and 10 months of age. The values presented are means±SD of n=10 mice. * P<0.05 compared to the wild-type (WT) group; # P<0.05 compared to the APP/PS1 group, as determined by analysis of variance (ANOVA), followed by the Tukey HSD test. Tables
Table 1. Sequences of the primers used to detect the APP and PS1 genes.Table 2. The content of MDA and activities of SOD and GSH-Px in the brains of mice at 6 months of age.Table 3. The content of MDA and activities of SOD and GSH-Px in the brains of mice at 10 months of age.Table 4. The content of MDA and activities of SOD and GSH-Px in the serum of mice at 6 months of age.Table 5. The content of MDA and activities of SOD and GSH-Px in the serum of mice at 10 months of age.Table 1. Sequences of the primers used to detect the APP and PS1 genes.Table 2. The content of MDA and activities of SOD and GSH-Px in the brains of mice at 6 months of age.Table 3. The content of MDA and activities of SOD and GSH-Px in the brains of mice at 10 months of age.Table 4. The content of MDA and activities of SOD and GSH-Px in the serum of mice at 6 months of age.Table 5. The content of MDA and activities of SOD and GSH-Px in the serum of mice at 10 months of age. In Press
06 Mar 2024 : Clinical Research
Comparison of Outcomes between Single-Level and Double-Level Corpectomy in Thoracolumbar Reconstruction: A ...Med Sci Monit In Press; DOI: 10.12659/MSM.943797
21 Mar 2024 : Meta-Analysis
Economic Evaluation of COVID-19 Screening Tests and Surveillance Strategies in Low-Income, Middle-Income, a...Med Sci Monit In Press; DOI: 10.12659/MSM.943863
10 Apr 2024 : Clinical Research
Predicting Acute Cardiovascular Complications in COVID-19: Insights from a Specialized Cardiac Referral Dep...Med Sci Monit In Press; DOI: 10.12659/MSM.942612
06 Mar 2024 : Clinical Research
Enhanced Surgical Outcomes of Popliteal Cyst Excision: A Retrospective Study Comparing Arthroscopic Debride...Med Sci Monit In Press; DOI: 10.12659/MSM.941102
Most Viewed Current Articles
17 Jan 2024 : Review article
Vaccination Guidelines for Pregnant Women: Addressing COVID-19 and the Omicron VariantDOI :10.12659/MSM.942799
Med Sci Monit 2024; 30:e942799
14 Dec 2022 : Clinical Research
Prevalence and Variability of Allergen-Specific Immunoglobulin E in Patients with Elevated Tryptase LevelsDOI :10.12659/MSM.937990
Med Sci Monit 2022; 28:e937990
16 May 2023 : Clinical Research
Electrophysiological Testing for an Auditory Processing Disorder and Reading Performance in 54 School Stude...DOI :10.12659/MSM.940387
Med Sci Monit 2023; 29:e940387
01 Jan 2022 : Editorial
Editorial: Current Status of Oral Antiviral Drug Treatments for SARS-CoV-2 Infection in Non-Hospitalized Pa...DOI :10.12659/MSM.935952
Med Sci Monit 2022; 28:e935952