18 September 2020: Clinical Research
TNNC1 Reduced Gemcitabine Sensitivity of Nonsmall-Cell Lung Cancer by Increasing Autophagy
Xian Ye1ABE, Guanghui Xie1D, Zhijian Liu1ABCDEFG, Jun Tang1F, Mingyuan Cui1FG, Chenbin Wang1DE, Chi Guo1CD, Jianfeng Tang1ABC*DOI: 10.12659/MSM.922703
Med Sci Monit 2020; 26:e922703






Abstract
BACKGROUND: As we know, chemotherapy resistance is a critical factor leading to recurrence and metastasis of nonsmall-cell lung cancer (NSCLC). To clarify the key target and potential mechanism of resistance to gemcitabine (GEM) in NSCLC, we selected Gene Expression Omnibus Data Set and statistically analyzed a parent cell group and a GEM-resistant cell group. Results showed that the expression of troponin C1, slow skeletal and cardiac type (TNNC1) in GEM-resistant cells was higher than in parent cells, which implies that TNNC1 was associated with GEM resistance in lung cancer cells.
MATERIAL AND METHODS: TNNC1 expression level was detected by reverse transcription-quantitative polymerase chain reaction or western blot in GEM-resistant patient serum and cell lines. It could reduce or increase autophagy response and GEM resistance accordingly by inhibition of the short interfering ribonucleic acid or by forced overexpression of TNNC1 viruses in A549 cell line and GEM-resistant cell line (A549/GemR) respectively. Blocking autophagy with 3-methyladenine increased the sensitivity of chemotherapy confirmed by flow cytometry and microtubule-associated protein 1A/1B – light chain 3 punctate assay. What’s more, in a loss-of-function model, silencing of forkhead box 03 (FOXO3) in A549/GemR cells could rescue the autophagy weakened by TNNC1.
RESULTS: TNNC1 promoted GEM chemoresistance of NSCLC by activating cytoprotective autophagy, regulated negatively by FOXO3. This research may provide a completely new strategy for NSCLC treatment.
CONCLUSIONS: Targeting the TNNC1/FOXO3 signaling pathway in NSCLC may be a novel strategy to combat GEM resistance.
Keywords: Forkhead Transcription Factors, Troponin C, A549 cells, Deoxycytidine, Neoplasm Proteins
Background
Lung cancer is one of the most prevalent malignant tumors in the world, and also the leading cause of cancer-related death [1] It is divided into two categories according to histological and clinical characteristics: small-cell lung cancer and nonsmall-cell lung cancer (NSCLC), with the latter accounting for 85–90% [2]. Though there has been significant progress in diagnosis and treatment in recent years, the overall 5-year survival rate for patients with NSCLC is still meager [3].
Chemotherapy is the most basic and effective therapy for NSCLC patients, especially for patients unable to undergo surgery. Gemcitabine (GEM) is a new pyrimidine antineoplastic drug mainly used for solid tumor therapy, including those in NSCLC [4]. Being a first-line therapeutic scheme for locally advanced-stage (stage III) and metastatic-stage (stage IV) NSCLC, it is used to treat NSCLC, with an effective rate of 20–30% [5,6]. Howeverr, its curative effect for lung cancer is restricted by intrinsic resistance, adaptive resistance, and acquired resistance; cancer cells can escape from the cytotoxicity of chemotherapeutic drugs by affecting cell cycle, cell apoptosis, or intracellular drug accumulation [7]. Presently, the exact molecular mechanism of resistance to GEM is not fully understood. Further study of this process will be useful to develop and improve cancer therapy strategies, and will also help to improve clinical efficacy.
Autophagy is a highly conserved lysosome degradation pathway that renews energy and maintains cell homeostasis through the degradation and recycling of proteins and damaged organelles [8]. As a self-protection mechanism used widely under stress conditions in eukaryotic cells, autophagy plays a vital role in cell survival, differentiation, aging, inflammation, and tumorigenesis [9,10]. Accumulated evidence shows that chemotherapy usually activates survival autophagy, which can reduce the apoptosis of cancer cells and then enable them to overcome the cytotoxicity or other stress-induced by therapy [11,12]. In NSCLC, autophagy intimately participated in the protective mechanism of resistance to chemotherapeutic agents. Pharmacologically or genetically induced autophagy inhibition can lead to sensitivity to chemotherapeutic agents; on the contrary, chemotherapy resistance appeared by its activation [13–15].
To clarify the key targets and potential mechanism of resistance to GEM in NSCLC, we selected Gene Expression Omnibus (GEO) Data Set (GSE6914) and statistically analyzed the parent cell group and the GEM-resistant cell group, and also conducted cluster analysis on messenger ribonucleic acid (mRNA), which showed the most significant difference between the two groups (|log2 fold change [FC]| >2,
Material and Methods
CELL CULTURE AND TREATMENT:
A549 (human NSCLC cell) and its GEM-resistant cells (A549/GemR) were provided by Procell Life Science & Technology Co., Ltd. (Wuhan, China). The cells were maintained in Ham’s F-12K media (Sigma, USA) with 10% fetal bovine serum (Gibco, USA) in an incubator with 5% CO2 at 37°C.
3-Methyladenine (3-MA), rapamycin (Rapa), and GEM were purchased from Selleck Chemicals (Shanghai, China). For drug handling, A549 cells were cultivated in 5 μM GEM, whereas A549/GemR cells were grown with 40 μM GEM. The concentration and processing time of 3-MA was 10 mM for 4 h, and Rapa was 10 μM for 4 h.
GENE SILENCED AND OVEREXPRESSED LENTIVIRUS PACKAGING:
Short interfering (si)RNA of TNNC1 and FOXO3 were devised and purchased from Thermo Fisher (TNNC1 sssay ID: 12778, 12868; 12955; FOXO3 assay ID: 41734, 41710, 115209, USA). To construct gene deleted-function cell models, TNNC1 and FOXO3 were designed with three siRNA sequences and transfected in A549/GemR cells using lipofectamine 2000 (Life Technologies, USA). The best interference efficiency in A549/GemR cells was confirmed by reverse tracscription-quantitative polymerase chain reaction (RT-qPCR). Functional assays were executed after 48 h of transfection. FOXO3 and TNNC1 were loaded into pCDNA3.1 vector to construct a FOXO3 overexpression plasmid and a TNNC1 overexpression plasmid respectively. TNNC1-overexpressed lentivirus was constructed and packaged by Genechem (Shanghai, China). The relative infection and transfection were conducted according to the manufacturer’s instructions.
SERUM COLLECTION:
A total of 20 primary lung adenocarcinoma (LUAD) blood specimens was selected. These patients were initially treated and had undergone GEM chemotherapy at the Centeral Hospital of Yongzhou under the procedures approved by the Ethics Committee for Clinical Trial of the Centeral Hospital of Yongzhou (Hunan, China). Ten of them developed drug resistance after taking GEM for three courses of treatment. In contrast, another 10 patients were sensitive to GEM. Fasting blood samples (10 mL) were obtained from each of the patients using serum separation tubes (BD, USA). The blood samples were transported within 30 min of collection and then centrifuged at 1900
CELL VIABILITY ASSAY:
To detect the sensitivity of GEM in A549/GemR cells and A549 cells, we incubated them with various concentrations of GEM. Different groups of different model cells were seeded into 96-well plates (approximately 1000 cells per well), and 10% 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) solution was added for about 4 h for each well. Then, cell proliferation viability for each well was measured on the basis of optical density measurements obtained at 490 nm. The experiment was repeated five times for each group. The negative control group was set as normal 100% survival.
RNA ISOLATION AND RT-QPCR:
Total RNA was collected from the cultured cells using TRIzol reagent (Invitrogen, USA) according to the manufacturer’s instructions. The expression of TNNC1 and FOXO3 mRNA was quantified by RT-qPCR analysis using SYBR Green PCR kit (Invitrogen). The following were the thermocycling conditions: predenaturation at 95°C for 15 min; then 95°C for 10 s for 45 cycles, 60°C for 20 s, and 72°C for 60 s with an amplification fragment of 122 base pairs (bp) for TNNC1 and 109 bp for FOXO3. The results were normalized to β-actin using the 2−ΔΔCq method to calculate the differential gene expression. Each reaction was implemented in triplicate.
Primers of TNNC1, FOXO3, and β-actin were as follows:
WESTERN BLOT ANALYSIS:
Cells were washed twice with precooled phosphate-buffered saline (PBS) and then ruptured with radioimmunoprecipitation assay lysis buffer (Beyotime, P0013B, Shanghai) supplement extracted with complete 5 mM ethylenediaminetetraacetic acid-free cocktail protease inhibitor. Cell extracts were centrifuged for 20 min at 10 000
APOPTOSIS ANALYSIS:
Annexin V-fluorescein isothiocyanate (FITC)/propidium iodide (PI) staining apoptosis detection kit (BD Biosciences) was used to detect apoptosis. A549 and A549/GemR model cells were cultured in six-well plates and pretreated with GEM or 3-MA and Rapa respectively. Cells were harvested in PBS, washed three times, and then resuspended in Annexin V-FITC/PI to stain cells at 4°C in darkness. The apoptotic statistical population was determined using FlowJo software.
IDENTIFICATION OF AUTOPHAGY:
A549 and A549/GemR model cells were transiently infected with AAv-mRFP-GFP-LC3 (Genechem). After incubation for 24 h, A549 and A549/GemR model cells were added into 5 μM or 40 μM GEM respectively for another 24 h. Finally, the GFP-LC3 punctate structures were visualized at ×200 using a fluorescence microscope (E3I-630, MOTIC, USA).
DUAL-LUCIFERASE REPORTER ASSAY:
To confirm our results that FOXO3 negatively regulated TNNC1, 1220 bp of the TNNC1 promoter region upstream sequence (1220 bp to 256 bp) from the starting codon was loaded to pGL3 luciferase reporter vector (Promega, USA) to construct a pGL3-TNNC1 promoter plasmid. The TNNC1 promoter sequence (965 bp) was synthesized by General Biosynthesis (Anhui, China). A549 cells were cotransfected with different reporter vectors using lipofectamine 2000 (Invitrogen): pGL3 basic vector cotransfected with pCDNA3.1 vector; pGL3-basic cotransfected with pCDNA3.1-FOXO3 (FOXO3); pGL3-TNNC1(TNNC1) cotransfected with pCDNA3.1; pGL3-TNNC1 cotransfected with pCDNA3.1-FOXO3. After 48 h, luciferase activity was detected through the dual luciferase reporter assay system (Promega). Experiments for each group were repeated three times.
STATISTICAL ANALYSIS:
GraphPad prism 7.0 software was used for statistical analysis. Differences between two groups were analyzed by Student’s
Results
TNNC1 WAS UPREGULATED MARKEDLY IN GEM-RESISTANT NSCLC CELLS AND SERUM OF PATIENTS:
To search the related GEM-resistant genes in NSCLC, we researched the GSE6914 first. We seeded out lots of potential mRNAs with obvious differences between GEM-resistant Calu3 cells (GSE159359-GSE15962) and Calu3 cells (GSE159355-GSE15958) (log FC>1.0 or <−1.0, P<0.05). TNNC1 was found as a significantly upregulated gene in GEM-resistant Calu3 cells (Calu3-GemR) compared with Calu3 cells (Calu3-parental) (log FC=3.445, P=0.000352, Figure 1A). Moreover, TNNC1was evidently increased more in the tumor tissues (tumor) than in paracancerous tissues (nomal) in LUAD (data sources GEPIA) (Figure 1B), which revealed that TNNC1 might have an association with NSCLC for occurrence and development. To further prove these results, TNNC1 was detected again in serum samples of 10 patients identified clinically as GEM resistant (resistance) and 10 patients identified clinically as GEM sensitive (sensitive) respectively. RT-qPCR showed that the TNNC1 expression was obviously stronger in Gem-resistant samples than in sensitive samples (Figure 1C). In addition, RT-qPCR and western blot were used to check TNNC1 in A549/GemR cells and A549 parent cells. The accordant conclusion was that TNNC1 upregulation in A549/GemR cells matched with A549 cells (Figure 1D, 1E). These results indicat that TNNC1 might play an important role in abating the effect of GEM in NSCLC.
TNNC1 OVEREXPRESSION COULD ENHANCE GEM SENSITIVITY IN A549 CELLS:
To research the role of TNNC1 in resistance of NSCLC to GEM, the TNNC1 overexpression model was established in A549 via infecting TNNC1 overexpression lentivirus (Lv-TNNC1), whereas the cell model of loss of function (siRNA-TNNC1) was established via transfecting TNNC1 siRNA in A549/GemR cells (Figure 2A). RT-qPCR was performed to verify that the TNNC1 functional model cells were obtained successfully (Figure 2B, 2C). Autophagy-related proteins LC3B and P62 were tested in A549 and A549/GemR model cells by western blot (Figure 2D). Protein levels of LC3II were increased obviously but P62 were decreased by TNNC1 overexpression. Concurrently, knockdown of TNNC1 (siRNA-TNNC1) in A549/GemR cells significantly reduced the expression of LC3BII. To choose the appropriate GEM concentration and incubation time, the cell proliferation ability in the A549 and A549/GemR model cells was analyzed by MTT after GEM treatment (Figure 2E). A549 cells were incubated with GEM at 0.05, 1.5, 5, 10, and 20 μM, whereas A549/GemR cells were incubated with GEM at 5, 10, 20, 40, and 80 μM. After incubation of both for 24 h, GEM (5, 10, and 20 μM) treatment significantly affected cell survival in A549 model cells. Meanwhile, GEM (40 and 80 μM) treatment significantly affected cell survival in A549/GemR model cells. Therefore, A549 model cells were incubated with 5 μM GEM, and A549/GemR model cells were incubated with 40 μM GEM. Then the time course of cell viability was assessed. Cell survival was significantly reduced within 24 h. As shown in Figure 2F, Lv-TNNC1 reduced the GEM-induced cell apoptosis evidently compared with the group infected with negative control lentivirus (Lv-NC) in A549 model cells treated with 5 μM GEM. In the meantime, in A549/GemR model cells treated with 40 μM GEM, siRNA-TNNC1 enhanced the GEM-induced cell apoptosis obviously compared with the group transfected with negative control siRNA (siRNA-NC). A549 and A549/GemR model cells were infected by AAv-mRFP-GFP-LC3 virus for 48 h. Before observation and imaging, cells were treated with 5 μM or 40 μM GEM for 24 h, respectively (Figure 2G). We found that RFP-LC3 puncta increased significantly in Lv-TNNC1 infection group compared with Lv-NC infection group in A549 cells treated with GEM 4 μM for 24 h. RFP-LC3 puncta were decreased in the group transfected with siRNA-TNNC1 compared with siRNA-NC in the A549/GemR cells treated with GEM at 40 μM for 24 h. Overall, all these results demonstrated that TNNC1 attenuated the GEM sensitivity of A549 cells. On the other hand, the inhibition of TNNC1 could enhance the sensitivity of A549/GemR cells to GEM.
TNNC1-PROMOTED GEM-INDUCED AUTOPHAGY PROTECTED A549 CELLS FROM APOPTOSIS:
On the basis of previous research results, we considered whether TNNC1 attenuated the GEM sensitivity of A549 cells through autophagy regulation. To confirm that TNNC1-promoted GEM led to survival autophagy that further protected A549 cells from apoptosis, we introduced an inhibitor (3-MA, 10 mM) of autophagy and Rapa (10 μM), the autophagy agonist, to A549 cells and A549/GemR cells for 4 h before analysis. At the same time, GEM was added at 5 μM to A549 cells and at 40 μM to A549/GemR cells, both for 24 h. Cell proliferation and viability were checked by MTT assay (Figure 3A, 3B). Meanwhile, western blot was used to study LC3B and P62 (Figure 3C) to verify the effect of 3-MA and Rapa on autophagy. Cell apoptosis was tested by flow cytometry with annexin V-FITC/PI double staining (Figure 3D). These results showed that suppressed autophagy enhanced cell apoptosis induced by GEM in A549 cells, in which TNNC1 was overexpressed. At the same time, autophagy activation enhanced the cell survival rate further by reducing GEM-induced cell apoptosis in TNNC1-depleted A549/GemR cells. We reviewed that TNNC1 promoted pro-survival autophagy induced by GEM to protect A549 cells from apoptotic death.
TNNC1 WAS NEGATIVELY REGULATED BY FOXO3:
Next, we tried to find out which transcription factors regulated TNNC1 expression. The program JASPAR was used to find the potential transcription factor for TNNC1, which was determined to be FOXO3 (Figure 4A). We tested the hypothesis that FOXO3 is a potential transcriptional suppressor for TNNC1 by the following experiments. First, we determined that FOXO3 can reduce the expression of TNNC1. A549 cells were directly transfected with an empty control vector plasmid in combination with a scrambled oligoucleotide (scramble) and FOXO3 overexpression plasmid (FOXO3-OVER). Western blots and RT-qPCR showed that FOXO3-OVER led to a significant downregulation of TNNC1 (Figure 4B–4D). Luciferase activity assay confirmed the interaction between TNNC1 promoter and FOXO3 (Figure 4E). These results revealed that FOXO3 overexpression weakened the luciferase activity of the reporter plasmid containing TNNC1 promoter compared with the empty vector (P<0.001). These showed that FOXO3 bound with TNNC1 promoter and further inhibited TNNC1 transcription.
TNNC1 PROMOTED AUTOPHAGY OF A549 CELLS AND AFFECTED GEM SENSITIVITY (TRANSCRIPTION REGULATED NEGATIVELY BY FOXO3):
Our previous research results showed that TNNC1 promoted autophagy of A549 cells and affected GEM sensitivity. Moreover, TNNC1 was negatively transcription regulated by FOXO3. Further analyses revealed that TNNC1 was regulated by FOXO3 transcription and further affected autophagy and GEM sensitivity of A549 cells. First, we established the overexpression or interference models for TNNC1 and FOXO3. In the TNNC1 overexpression model, A549 cells were transfected with FOXO3 plasmid (TNNC1+FOXO3) for 48 h; at the same time, TNNC1 overexpression model cells (TNNC1) were used for the control group. Then, A549/GemR cells were split into two groups. One group was transfected with TNNC1 siRNA for 48 h and the other group was co-transfected with TNNC1 siRNA+FOXO3 siRNA for 48 h. All of the A549 and A549/GemR cells were incubated with GEM for 24 h before being tested. MTT arrays demonstrated that the ability of cell proliferation weakened by TNNC1 lost could be restored after FOXO3 silencing (Figure 5A). Consistent with this, the proliferation activity of A549/GemR cells induced by TNNC1 could be repressed by FOXO3 (Figure 5B). The results of western blot analyzed the autophagy-related proteins LC3B and P62 and confirmed that FOXO3 could abate the autophagy induced by TNNC1. Meanwhile, FOXO3 siRNA restored the autophagy that was weakened by TNNC1 siRNA in A549/GemR (Figure 5C). Next, flow cytometry with annexin V-FITC/PI double staining was used to analysis A549 and A549/GemR cell apoptosis rate (Figure 5D). A549 and A549/GemR cells of different groups were infected with AAv-mRFP-GFP-LC3 lentivirus for 48 h and later GFP-LC3 puncta accumulation was imaged under a fluorescence microscope (Figure 5E). These findings suggested that FOXO3, as one of the functional upstream effectors of TNNC1, negatively transcription regulated TNNC1 on GEM sensitivity of A549 cells.
Discussion
Although chemotherapy is the first treatment for advanced lung cancer, drug resistance is a crucial barrier to prolonging survival. Cancer cells may evade the cytotoxic effects of chemotherapeutic drugs by regulating the cell cycle, apoptosis, or accumulation of intracellular drugs. In this present study, we revealed a mechanism by which NSCLC cell evasion of chemotherapeutic drugs (GEM) is mediated by autophagy associated with TNNC1.
To find the critical targets for GEM resistance of NSCLC, we performed a query analysis on the GEO online database; the results showed that TNNC1 was significantly upregulated in GEM-resistant cells compared with parental cells (Figure 1A), suggesting that TNNC1 may be related to the development of chemotherapy resistance. The outcomes of the experiment showed that TNNC1 was downregulated and compared clinically sensitive samples and A549 parent cells with corresponding resistance groups (Figure 1C–1E), which is accord with the results of GEO data analysis. The present finding suggested that TNNC1 was closely associated with GEM resistance in NSCLC. TNNC1 contains calcium-binding subunits, which are often unformed in muscle cells, that promote the interaction of actin and myosin in muscle cells and promote the formation of stress fibers [16,17]. In nonmuscle cells, TNNC1 did not act as a structural protein but as a regulatory protein for cell motility, cytoplasmic flow, and cell division. For example, increasing the expression of TNNC1 in epithelial ovarian cancer cells governed the movement and invasiveness of the cancer cells through cytoskeletal reorganization, associated with poor survival [18,24]. It was not reported how TNNC1 affects GEM resistance. Previous investigations had illustrated that enhanced autophagy was an essential mechanism of GEM resistance in cancer cells. Autophagy plays a protective role in cancer cells, preventing them from entering the apoptotic pathway after stimulation with GEM, whereas blocking autophagy can enhance the tumoricidal effect of GEM. TNNC1 contains a calcium-binding subunit, and Ca2+ acts as an essential regulator of autophagy [25–27]. Whether TNNC1 can regulate autophagy has not been well characterized. So far, we speculated that TNNC1 may affect GEM resistance by regulating autophagy.
As expected, our results first showed that autophagy was weakened by decreasing TNNC1 in A549/GemR cells (Figure 2D, 2G). It was proposed that increased TNNC1 expression might lead to enhanced autophagy and reduce GEM-induced apoptosis in the NSCLC cells, similarly to previous studies [13–15]. Moreover, we found that TNNC1-mediated apoptosis was effectively enhanced when 3-MA was used to inhibit autophagy, whereas apoptosis was attenuated when autophagy was activated by Rapa, which was demonstrated by apoptotic cell percentage (Figure 3D) and the expression of autophagy-related proteins (Figure 3C). Given the above, it was evident that TNNC1 regulated the sensitivity of lung cancer cells to GEM chemotherapy, at least in part, by autophagy. Further research is needed to research how TNNC1 regulates autophagy. This study did not address this issue because of time and funding constraints.
Nevertheless, the molecular mechanism of TNNC1 enhancement in GEM-resistant patients and cells is largely unknown. As documented, FOXO3 as a common transcription modulator of autophagy and tumor suppressor during cancer cell proliferation, invasion, metastasis, metabolism, and apoptosis [28,29]. It is worth mentioning that FOXO3 was downregulated in GEM-resistant A549 cells, which was the opposite of TNNC1 (Figure 1D, 1E). FOXO3 was predicted to be a potential transcription factor for the TNNC1 gene by online prediction analysis by JASPAR (Figure 4A). We speculated that FOXO3 was related to the expression of TNNC1. Next, we researched the regulating effect of FOXO3 for TNNC1-mediated autophagy as well as the GEM resistance of NSCLC. The dual-luciferase reporter assay confirmed that FOXO3 bound to the TNNC1 promoter region and inhibited TNNC1 transcription (Figure 4D). Therefore, we further studied the cell function model of loss and overexpression in A549/GemR or A549 cells respectively. These results showed that overexpression or inhibition of FOXO3 were able to weaken or rescue TNNC1-mediated autophagy, respectively (Figure 5C–5E). Our data suggested that FOXO3 regulated TNNC1-mediated autophagy, whereas it also participated in chemoresistance through transcription suppression of TNNC1. It was possible that other regulating genes of TNNC1 also contribute to the autophagy-mediated GEM resistance in NSCLC. Also, our present study highlighted the function of FOXO3 at the transcriptional level and uncovered the TNNC1 regulation axis in response to GEM treatment in NSCLC.
It should be noted that TNNC1 was not related to the prognosis of LUAD derived from the TCGA database (
In summary, we provided further evidence to support that activated TNNC1 in A549 cells could facilitate autophagy and protect cells from apoptosis, thereby impairing the sensitivity of A549 cells to GEM chemotherapy, although the transcriptional regulator FOXO3 can be combined with the promoter region of the TNNC1 gene and negatively regulate TNNC1. Collectively, we deduced that TNNC1-mediated GEM resistance associated with autophagy was negatively regulated by FOXO3 in NSCLC cells. These findings had an insight deeply into molecular mechanisms underlying GEM resistance of NSCLC. Thus, targeting TNNC1-mediated autophagy might be a valid strategy to find novel therapeutic agents for NSCLC.
Conclusions
To research the mechanism of drug resistance in NSCLC, we obtained data on TNNC1 by analyzing the existing database GSE6914. We provided significant evidence that TNNC1 could enhance autophagy and further reduce the chemosensitivity of A549 cells to GEM. However, how TNNC1 regulated autophagy still needs to be researched. It was also demonstrated that FOXO3 could be used as the upstream negative regulatory factor of TNNC1. The chemosensitivity of TNNC1 to GEM was regulated by FOXO3. In the future, we will perform research on the regulation mechanism of the TNNC1/FOXO3 signaling pathway.
Figures
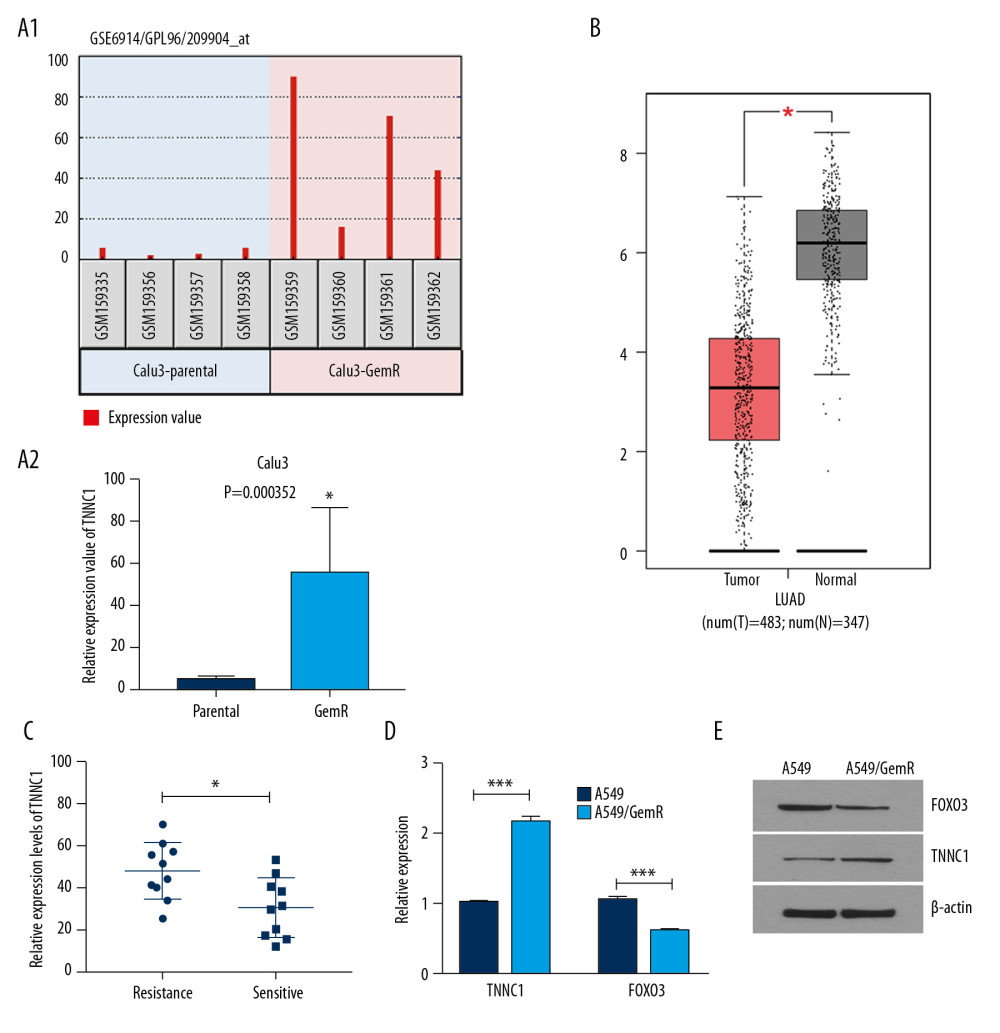 Figure 1. Troponin C1, slow skeletal and cardiac type (TNNC1) was significantly upregulated in gemcitabine (GEM)-resistant nonsmall-cell lung cancer cells and serum of the patient. (A1, A2) TNNC1 was significantly upregulated in GEM-resistant Calu3 cells (Calu3-GemR) compared with the parental Calu3 cells (Calu3-parental) by analyzing the Gene Expression Omnibus Data Set log fold change >1.0 or <−1.0, P<0.05). (B) TNNC1 was significantly increased in lung adenocarcinoma cancer tissues (Tumor) compared with adjacent tissues (Normal) (data sources Gene Expression Profiling Interactive Analysis database). (C) Expression of TNNC1 in clinical serum samples using reverse tracscription-quantitative polymerase chain reaction (RT-qPCR). Resistance group vs. sensitive group; data presented as mean±SD, * P<0.05 (D, E). Relative expression of TNNC1 and forkhead box 03 (FOXO3) in A549/GemR cells and A549 parental cells detected by RT-qPCR (D) and western blot (E) respectively (*** P<0.001).
Figure 1. Troponin C1, slow skeletal and cardiac type (TNNC1) was significantly upregulated in gemcitabine (GEM)-resistant nonsmall-cell lung cancer cells and serum of the patient. (A1, A2) TNNC1 was significantly upregulated in GEM-resistant Calu3 cells (Calu3-GemR) compared with the parental Calu3 cells (Calu3-parental) by analyzing the Gene Expression Omnibus Data Set log fold change >1.0 or <−1.0, P<0.05). (B) TNNC1 was significantly increased in lung adenocarcinoma cancer tissues (Tumor) compared with adjacent tissues (Normal) (data sources Gene Expression Profiling Interactive Analysis database). (C) Expression of TNNC1 in clinical serum samples using reverse tracscription-quantitative polymerase chain reaction (RT-qPCR). Resistance group vs. sensitive group; data presented as mean±SD, * P<0.05 (D, E). Relative expression of TNNC1 and forkhead box 03 (FOXO3) in A549/GemR cells and A549 parental cells detected by RT-qPCR (D) and western blot (E) respectively (*** P<0.001). 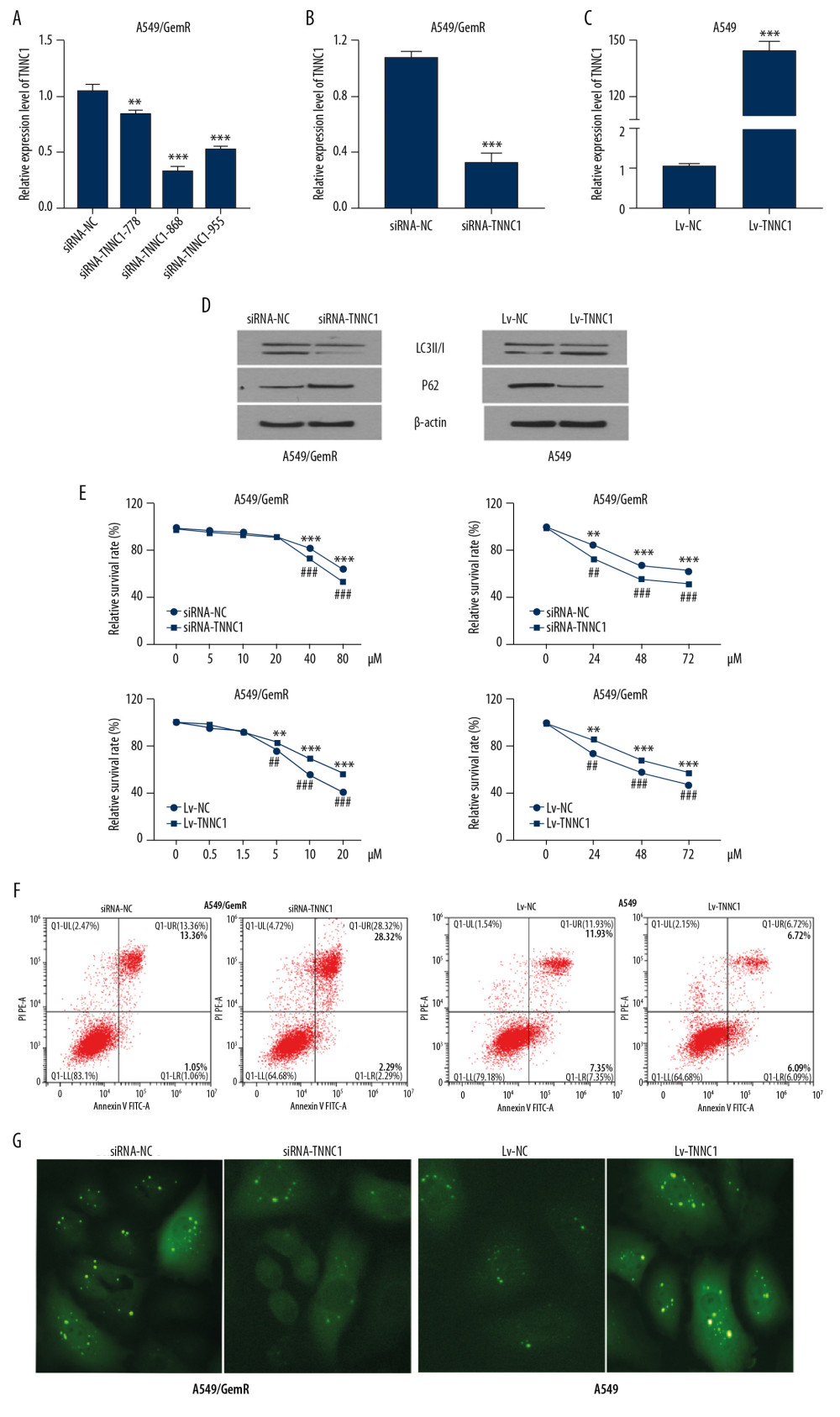 Figure 2. Troponin C1, slow skeletal and cardiac type (TNNC1) overexpression could enhance the sensitivity of A549 cells to gemcitabine (GEM). (A) Detection of the knockout effect of TNNC1. (B) TNNC1 lost model was successfully established in A549/GemR cells by transfecting TNNC1 short interfering ribonucleic acid (siRNA-TNNC1) and negative control RNA (siRNA-NC) respectively by reverse tracscription-quantitative polymerase chain reaction (RT-qPCR) (*** P<0.01). (C) Stable TNNC1-overexpressed cell line was successfully established in A549 cells by infecting with TNNC1 overexpression lentivirus (Lv-TNNC1) or negative control lentivirus (Lv-NC) respectively through RT-qPCR assay (*** P<0.01). (D) Western blot assay was used to evaluate the expression of autophagy-related proteins in A549 and A549/GemR model cells. (E) A549 cells were incubated with increasing concentrations of GEM (0–20 μM) for 24 h; meanwhile A549/GemR cells were incubated with increasing concentrations of GEM (0–80 μM) for 24 h. Cell viability was assessed by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT). A549 were incubated with 5 μM GEM, whereas A549/GemR were incubated with 40 μM GEM. Then cell viability was determined at 24, 48, and 72 h by MTT assay. The results were shown as mean±SD of four independent experiments. (** P<0.01, *** P<0.001, ## P<0.01, ### P<0.001). (F) Flow cytometry with annexin V-fluorescein isothiocyanate/propidium iodide double staining detected apoptosis in A549 and A549/GemR model cells after GEM treatment respectively with 5 μM and 40 μM for 24 h. (G) LC3 puncta accumulation in A549 and A549/GemR model cells after GEM treatment with 5 μM and 40 μM for 24 h, respectively. The original magnification was ×200.
Figure 2. Troponin C1, slow skeletal and cardiac type (TNNC1) overexpression could enhance the sensitivity of A549 cells to gemcitabine (GEM). (A) Detection of the knockout effect of TNNC1. (B) TNNC1 lost model was successfully established in A549/GemR cells by transfecting TNNC1 short interfering ribonucleic acid (siRNA-TNNC1) and negative control RNA (siRNA-NC) respectively by reverse tracscription-quantitative polymerase chain reaction (RT-qPCR) (*** P<0.01). (C) Stable TNNC1-overexpressed cell line was successfully established in A549 cells by infecting with TNNC1 overexpression lentivirus (Lv-TNNC1) or negative control lentivirus (Lv-NC) respectively through RT-qPCR assay (*** P<0.01). (D) Western blot assay was used to evaluate the expression of autophagy-related proteins in A549 and A549/GemR model cells. (E) A549 cells were incubated with increasing concentrations of GEM (0–20 μM) for 24 h; meanwhile A549/GemR cells were incubated with increasing concentrations of GEM (0–80 μM) for 24 h. Cell viability was assessed by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT). A549 were incubated with 5 μM GEM, whereas A549/GemR were incubated with 40 μM GEM. Then cell viability was determined at 24, 48, and 72 h by MTT assay. The results were shown as mean±SD of four independent experiments. (** P<0.01, *** P<0.001, ## P<0.01, ### P<0.001). (F) Flow cytometry with annexin V-fluorescein isothiocyanate/propidium iodide double staining detected apoptosis in A549 and A549/GemR model cells after GEM treatment respectively with 5 μM and 40 μM for 24 h. (G) LC3 puncta accumulation in A549 and A549/GemR model cells after GEM treatment with 5 μM and 40 μM for 24 h, respectively. The original magnification was ×200. 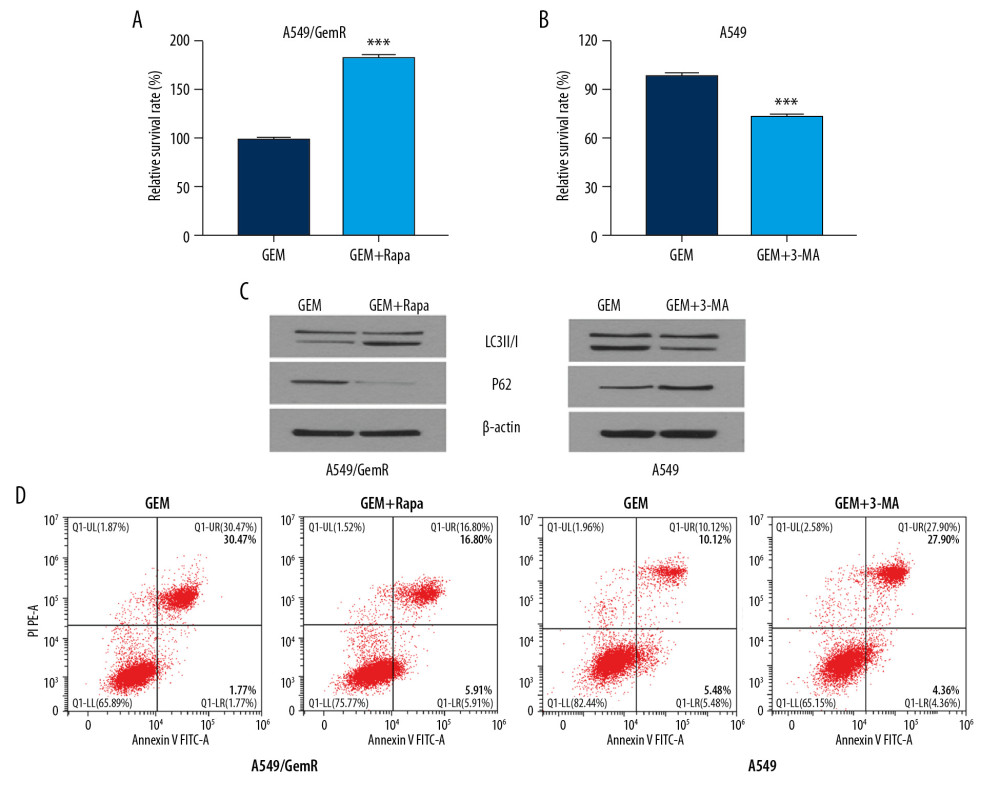 Figure 3. Troponin C1, slow skeletal and cardiac type (TNNC1) promoted gemcitabine (GEM)-induced autophagy and protected the nonsmall-cell lung cancer cells from apoptosis. (A, B) 3-Methyladenine (3-MA, 10 mM) and rapamycin (10 μM) were added to A549 and A549/GemR model cell cultures respectively for 4 h before GEM exposure. 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide was used to estimate the relative survival rate. The results are shown as mean±SD (*** P<0.001). (C) Western blot assay was used to evaluate the autophagy-related proteins LC3B and P62 in A549 and A549/GemR model cells after GEM treatment at 24 h and 3-MA (10 mM) or rapamycin(10 μM) was added at 4 h respectively. (D) Flow cytometry with annexin V–fluorescein isothiocyanate/propidium iodide double staining was used to detect cell apoptosis.
Figure 3. Troponin C1, slow skeletal and cardiac type (TNNC1) promoted gemcitabine (GEM)-induced autophagy and protected the nonsmall-cell lung cancer cells from apoptosis. (A, B) 3-Methyladenine (3-MA, 10 mM) and rapamycin (10 μM) were added to A549 and A549/GemR model cell cultures respectively for 4 h before GEM exposure. 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide was used to estimate the relative survival rate. The results are shown as mean±SD (*** P<0.001). (C) Western blot assay was used to evaluate the autophagy-related proteins LC3B and P62 in A549 and A549/GemR model cells after GEM treatment at 24 h and 3-MA (10 mM) or rapamycin(10 μM) was added at 4 h respectively. (D) Flow cytometry with annexin V–fluorescein isothiocyanate/propidium iodide double staining was used to detect cell apoptosis. 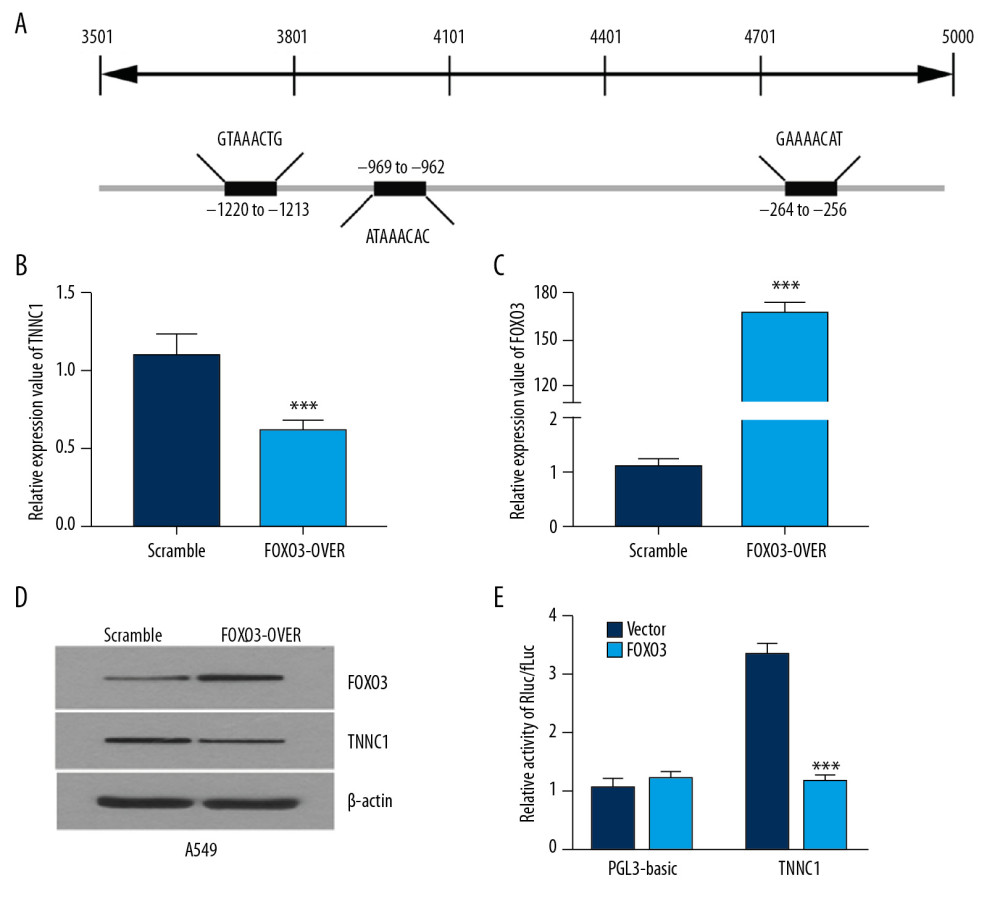 Figure 4. Troponin C1, slow skeletal and cardiac type (TNNC1) was negatively regulated by forkhead box 03 (FOXO3). (A) Schematic diagram of the FOXO3 binding sites on the promoter of TNNC1. The expression of TNNC1 in A549 cells transfected with an empty vector (Scramble) or FOXO3 overexpression plasmid (FOXO3-over) was determined by reverse tracscription-quantitative polymerase chain reaction (B, C) or western blot (D). Dual-luciferase reporter assay was used to analyze the TNNC1 promoter activity. The promoter constructs were cotransfected with empty vector (Vector) or FOXO3 overexpression plasmid (FOXO3). (E) Luciferase level was significantly attenuated after the cotransfection of TNNC1 promoter with FOXO3 plasmid compared with the vector plasmid.
Figure 4. Troponin C1, slow skeletal and cardiac type (TNNC1) was negatively regulated by forkhead box 03 (FOXO3). (A) Schematic diagram of the FOXO3 binding sites on the promoter of TNNC1. The expression of TNNC1 in A549 cells transfected with an empty vector (Scramble) or FOXO3 overexpression plasmid (FOXO3-over) was determined by reverse tracscription-quantitative polymerase chain reaction (B, C) or western blot (D). Dual-luciferase reporter assay was used to analyze the TNNC1 promoter activity. The promoter constructs were cotransfected with empty vector (Vector) or FOXO3 overexpression plasmid (FOXO3). (E) Luciferase level was significantly attenuated after the cotransfection of TNNC1 promoter with FOXO3 plasmid compared with the vector plasmid. 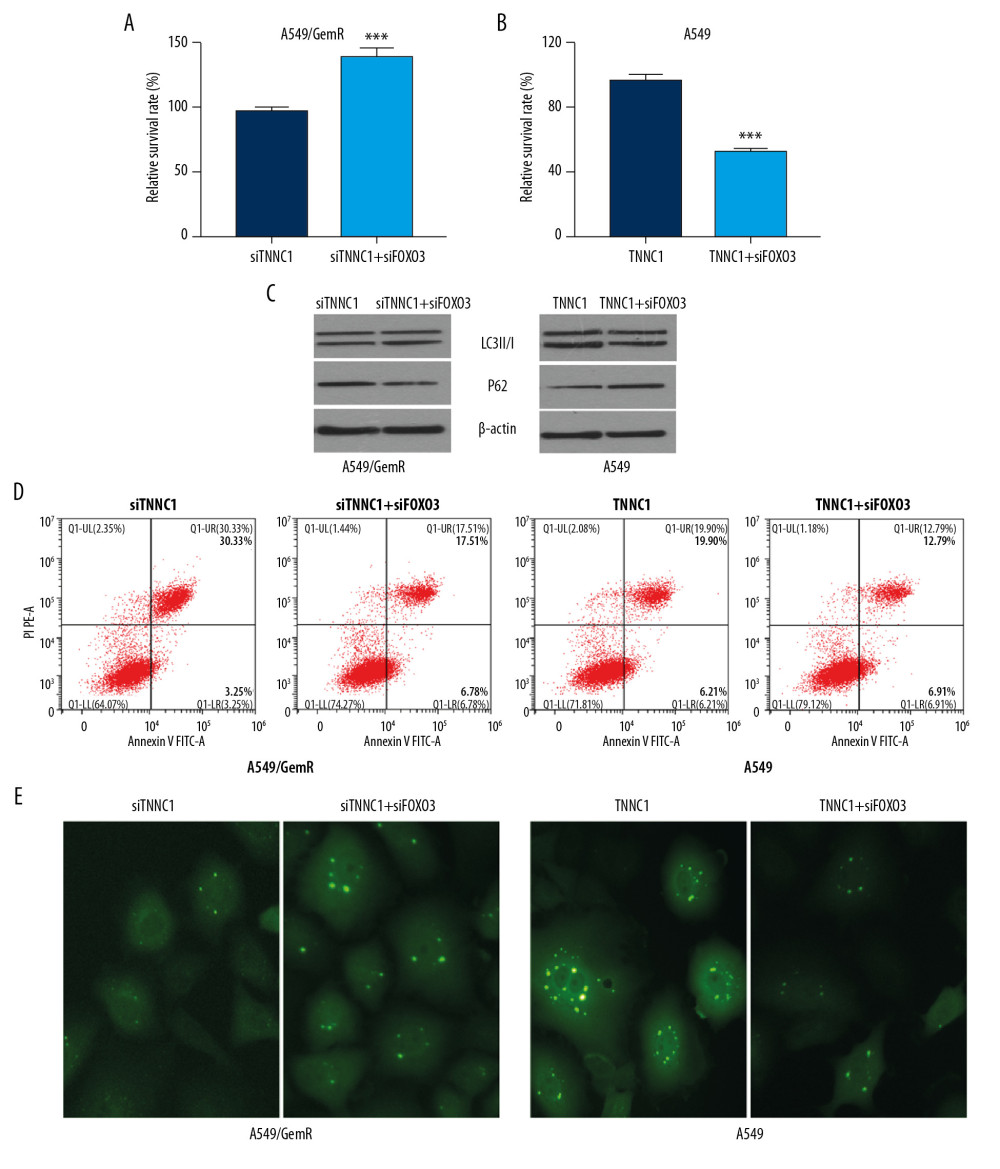 Figure 5. Troponin C1, slow skeletal and cardiac type (TNNC1) promoted autophagy of A549 cells and affected gemcitabine (GEM) sensitivity, which was negatively transcription regulated by forkhead box 03 (FOXO3). A549-overexpressed TNNC1-stable cells were transfected with FOXO3 overexpression plasmid (TNNC1+FOXO3) for 48 h, whereas A549-overexpressed TNNC1-stable cells (TNNC1) were used as controls. At the same time, A549/GemR cells were transfected with TNNC1 short interfering ribonucleic acid (siRNA) (siTNNC1) or TNNC1 siRNA plus FOXO3 siRNA (siTNNC1+siFOXO3) respectively for 48 h. Meanwhile, GEM was incubated for 24 h before testing. (A, B) Cell proliferation of A549 and A549/GemR was determined by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. The data were presented as the mean±SD (*** P<0.001). (C) Autophagy-related proteins LC3B and P62 were detected by western blot. (D) Apoptosis was investigated by flow cytometry. (E) GFP-LC3 punctate structures were observed and imaged using a fluorescence microscope in A549 and A549/GemR model cells after AAv-mRFP-GFP-LC3 viruses infected them. The original magnification was ×200.
Figure 5. Troponin C1, slow skeletal and cardiac type (TNNC1) promoted autophagy of A549 cells and affected gemcitabine (GEM) sensitivity, which was negatively transcription regulated by forkhead box 03 (FOXO3). A549-overexpressed TNNC1-stable cells were transfected with FOXO3 overexpression plasmid (TNNC1+FOXO3) for 48 h, whereas A549-overexpressed TNNC1-stable cells (TNNC1) were used as controls. At the same time, A549/GemR cells were transfected with TNNC1 short interfering ribonucleic acid (siRNA) (siTNNC1) or TNNC1 siRNA plus FOXO3 siRNA (siTNNC1+siFOXO3) respectively for 48 h. Meanwhile, GEM was incubated for 24 h before testing. (A, B) Cell proliferation of A549 and A549/GemR was determined by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. The data were presented as the mean±SD (*** P<0.001). (C) Autophagy-related proteins LC3B and P62 were detected by western blot. (D) Apoptosis was investigated by flow cytometry. (E) GFP-LC3 punctate structures were observed and imaged using a fluorescence microscope in A549 and A549/GemR model cells after AAv-mRFP-GFP-LC3 viruses infected them. The original magnification was ×200. References
1. Bray F, Ferlay J, Soerjomataram I, Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries: Cancer J Clin, 2018; 68; 394-424
2. Kuribayashi K, Funaguchi N, Nakano T, Chemotherapy for advanced non-small cell lung cancer with a focus on squamous cell carcinoma: J Cancer Res Ther, 2016; 12; 528-34
3. Wang H, Zhou Y, Liu Q, Prognostic value of SOX2, Cyclin D1, P53, and ki-67 in patients with esophageal squamous cell carcinoma: Onco Targets Ther, 2018; 11; 5171-81
4. Reck M, Heigener DF, Mok T, Management of non-small-cell lung cancer: Recent developments: Lancet, 2013; 382; 709-19
5. Wu YL, Zhou C, Hu CP, Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): An open-label, randomised phase 3 trial: Lancet Oncol, 2014; 15; 213-22
6. Thatcher N, Hirsch FR, Luft AV, Necitumumab plus gemcitabine and cisplatin versus gemcitabine and cisplatin alone as first-line therapy in patients with stage IV squamous non-small-cell lung cancer (SQUIRE): An open-label, randomised, controlled phase 3 trial: Lancet Oncol, 2015; 16; 763-74
7. Tokunaga Y, Liu D, Nakano J, Potent effect of adenoviral vector expressing short hairpin RNA targeting ribonucleotide reductase large subunit M1 on cell viability and chemotherapeutic sensitivity to gemcitabine in non-small cell lung cancer cells: Eur J Cancer, 2015; 51; 2480-89
8. Mizushima N, Autophagy: Process and function: Genes Dev, 2007; 21; 2861-73
9. Kaminskyy VO, Piskunova T, Zborovskaya IB, Suppression of basal autophagy reduces lung cancer cell proliferation and enhances caspase-dependent and -independent apoptosis by stimulating ROS formation: Autophagy, 2012; 8; 1032-44
10. Jiang GM, Tan Y, Wang H, The relationship between autophagy and the immune system and its applications for tumor immunotherapy: Mol Cancer, 2019; 18; 17
11. Yu T, Guo F, Yu Y, Fusobacterium nucleatum promotes chemoresistance to colorectal cancer by modulating autophagy: Cell, 2017; 170; 548-563.e16
12. Xu S, Wang P, Zhang J, Ai-lncRNA EGOT enhancing autophagy sensitizes paclitaxel cytotoxicity via upregulation of ITPR1 expression by RNA-RNA and RNA-protein interactions in human cancer: Mol Cancer, 2019; 18; 89
13. Meng J, Chang C, Chen Y, EGCG overcomes gefitinib resistance by inhibiting autophagy and augmenting cell death through targeting ERK phosphorylation in NSCLC: Onco Targets Ther, 2019; 12; 6033-43
14. Zhang L, Qiang P, Yu J, Identification of compound CA-5f as a novel late-stage autophagy inhibitor with potent anti-tumor effect against non-small cell lung cancer: Autophagy, 2019; 15; 391-406
15. Wu HM, Shao LJ, Jiang ZF, Liu RY, Gemcitabine-induced autophagy protects human lung cancer cells from apoptotic death: Lung, 2016; 194; 959-66
16. Tash JS, Means AR, Regulation of protein phosphorylation and motility of sperm by cyclic adenosine monophosphate and calcium: Biol Reprod, 1982; 26; 745-63
17. Li MX, Hwang PM, Structure and function of cardiac troponin C (TNNC1): Implications for heart failure, cardiomyopathies, and troponin modulating drugs: Gene, 2015; 571; 153-66
18. Leung CS, Yeung TL, Yip KP, Calcium-dependent FAK/CREB/TNNC1 signalling mediates the effect of stromal MFAP5 on ovarian cancer metastatic potential: Nat Commun, 2014; 5; 5092
19. Zhou J, Liao W, Yang J, FOXO3 induces FOXO1-dependent autophagy by activating the AKT1 signaling pathway: Autophagy, 2012; 8; 1712-23
20. Wen Q, Jiao X, Kuang F, FoxO3a inhibiting expression of EPS8 to prevent progression of NSCLC: A new negative loop of EGFR signaling: EBioMedicine, 2019; 40; 198-209
21. Cai J, Li R, Xu X, CK1alpha suppresses lung tumour growth by stabilizing PTEN and inducing autophagy: Nat Cell Biol, 2018; 20; 465-78
22. Yao S, Fan LY, Lam EW, The FOXO3-FOXM1 axis: A key cancer drug target and a modulator of cancer drug resistance: Semin Cancer Biol, 2018; 50; 77-89
23. Guan L, Zhang L, Gong Z, FoxO3 inactivation promotes human cholangiocarcinoma tumorigenesis and chemoresistance through Keap1-Nrf2 signaling: Hepatology, 2016; 63; 1914-27
24. Gahlmann R, Wade R, Gunning P, Kedes L, Differential expression of slow and fast skeletal muscle troponin C. Slow skeletal muscle troponin C is expressed in human fibroblasts: J Mol Biol, 1988; 201; 379-91
25. East DA, Campanella M, Ca2+ in quality control: An unresolved riddle critical to autophagy and mitophagy: Autophagy, 2013; 9; 1710-19
26. Vlahakis A, Lopez Muniozguren N, Powers T, Mitochondrial respiration links TOR complex 2 signaling to calcium regulation and autophagy: Autophagy, 2017; 13; 1256-57
27. Qu YQ, Gordillo-Martinez F, Law BYK, 2-Aminoethoxydiphenylborane sensitizes anti-tumor effect of bortezomib via suppression of calcium-mediated autophagy: Cell Death Dis, 2018; 9; 361
28. Qian Z, Ren L, Wu D, Overexpression of FoxO3a is associated with glioblastoma progression and predicts poor patient prognosis: Int J Cancer, 2017; 140; 2792-804
29. Fitzwalter BE, Thorburn A, FOXO3 links autophagy to apoptosis: Autophagy, 2018; 14; 1467-68
30. Yang X, Wu K, Li S, MFAP5 and TNNC1: Potential markers for predicting occult cervical lymphatic metastasis and prognosis in early stage tongue cancer: Oncotarget, 2017; 8; 2525-35
31. Mogensen J, Murphy RT, Shaw T, Severe disease expression of cardiac troponin C and T mutations in patients with idiopathic dilated cardiomyopathy: J Am Coll Cardiol, 2004; 44; 2033-40
Figures
Figure 1. Troponin C1, slow skeletal and cardiac type (TNNC1) was significantly upregulated in gemcitabine (GEM)-resistant nonsmall-cell lung cancer cells and serum of the patient. (A1, A2) TNNC1 was significantly upregulated in GEM-resistant Calu3 cells (Calu3-GemR) compared with the parental Calu3 cells (Calu3-parental) by analyzing the Gene Expression Omnibus Data Set log fold change >1.0 or <−1.0, P<0.05). (B) TNNC1 was significantly increased in lung adenocarcinoma cancer tissues (Tumor) compared with adjacent tissues (Normal) (data sources Gene Expression Profiling Interactive Analysis database). (C) Expression of TNNC1 in clinical serum samples using reverse tracscription-quantitative polymerase chain reaction (RT-qPCR). Resistance group vs. sensitive group; data presented as mean±SD, * P<0.05 (D, E). Relative expression of TNNC1 and forkhead box 03 (FOXO3) in A549/GemR cells and A549 parental cells detected by RT-qPCR (D) and western blot (E) respectively (*** P<0.001).Figure 2. Troponin C1, slow skeletal and cardiac type (TNNC1) overexpression could enhance the sensitivity of A549 cells to gemcitabine (GEM). (A) Detection of the knockout effect of TNNC1. (B) TNNC1 lost model was successfully established in A549/GemR cells by transfecting TNNC1 short interfering ribonucleic acid (siRNA-TNNC1) and negative control RNA (siRNA-NC) respectively by reverse tracscription-quantitative polymerase chain reaction (RT-qPCR) (*** P<0.01). (C) Stable TNNC1-overexpressed cell line was successfully established in A549 cells by infecting with TNNC1 overexpression lentivirus (Lv-TNNC1) or negative control lentivirus (Lv-NC) respectively through RT-qPCR assay (*** P<0.01). (D) Western blot assay was used to evaluate the expression of autophagy-related proteins in A549 and A549/GemR model cells. (E) A549 cells were incubated with increasing concentrations of GEM (0–20 μM) for 24 h; meanwhile A549/GemR cells were incubated with increasing concentrations of GEM (0–80 μM) for 24 h. Cell viability was assessed by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT). A549 were incubated with 5 μM GEM, whereas A549/GemR were incubated with 40 μM GEM. Then cell viability was determined at 24, 48, and 72 h by MTT assay. The results were shown as mean±SD of four independent experiments. (** P<0.01, *** P<0.001, ## P<0.01, ### P<0.001). (F) Flow cytometry with annexin V-fluorescein isothiocyanate/propidium iodide double staining detected apoptosis in A549 and A549/GemR model cells after GEM treatment respectively with 5 μM and 40 μM for 24 h. (G) LC3 puncta accumulation in A549 and A549/GemR model cells after GEM treatment with 5 μM and 40 μM for 24 h, respectively. The original magnification was ×200.Figure 3. Troponin C1, slow skeletal and cardiac type (TNNC1) promoted gemcitabine (GEM)-induced autophagy and protected the nonsmall-cell lung cancer cells from apoptosis. (A, B) 3-Methyladenine (3-MA, 10 mM) and rapamycin (10 μM) were added to A549 and A549/GemR model cell cultures respectively for 4 h before GEM exposure. 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide was used to estimate the relative survival rate. The results are shown as mean±SD (*** P<0.001). (C) Western blot assay was used to evaluate the autophagy-related proteins LC3B and P62 in A549 and A549/GemR model cells after GEM treatment at 24 h and 3-MA (10 mM) or rapamycin(10 μM) was added at 4 h respectively. (D) Flow cytometry with annexin V–fluorescein isothiocyanate/propidium iodide double staining was used to detect cell apoptosis.Figure 4. Troponin C1, slow skeletal and cardiac type (TNNC1) was negatively regulated by forkhead box 03 (FOXO3). (A) Schematic diagram of the FOXO3 binding sites on the promoter of TNNC1. The expression of TNNC1 in A549 cells transfected with an empty vector (Scramble) or FOXO3 overexpression plasmid (FOXO3-over) was determined by reverse tracscription-quantitative polymerase chain reaction (B, C) or western blot (D). Dual-luciferase reporter assay was used to analyze the TNNC1 promoter activity. The promoter constructs were cotransfected with empty vector (Vector) or FOXO3 overexpression plasmid (FOXO3). (E) Luciferase level was significantly attenuated after the cotransfection of TNNC1 promoter with FOXO3 plasmid compared with the vector plasmid.Figure 5. Troponin C1, slow skeletal and cardiac type (TNNC1) promoted autophagy of A549 cells and affected gemcitabine (GEM) sensitivity, which was negatively transcription regulated by forkhead box 03 (FOXO3). A549-overexpressed TNNC1-stable cells were transfected with FOXO3 overexpression plasmid (TNNC1+FOXO3) for 48 h, whereas A549-overexpressed TNNC1-stable cells (TNNC1) were used as controls. At the same time, A549/GemR cells were transfected with TNNC1 short interfering ribonucleic acid (siRNA) (siTNNC1) or TNNC1 siRNA plus FOXO3 siRNA (siTNNC1+siFOXO3) respectively for 48 h. Meanwhile, GEM was incubated for 24 h before testing. (A, B) Cell proliferation of A549 and A549/GemR was determined by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. The data were presented as the mean±SD (*** P<0.001). (C) Autophagy-related proteins LC3B and P62 were detected by western blot. (D) Apoptosis was investigated by flow cytometry. (E) GFP-LC3 punctate structures were observed and imaged using a fluorescence microscope in A549 and A549/GemR model cells after AAv-mRFP-GFP-LC3 viruses infected them. The original magnification was ×200. In Press
Clinical Research
Effects of Single-Bout Endurance Exercise Intensity on Peripheral Neurotrophic Factors in Patients With Isc...Med Sci Monit In Press; DOI: 10.12659/MSM.952089
Review article
Anisodus tanguticus in Cancer Research: A Review of Traditional Use, Phytochemistry, Extraction Methods, an...Med Sci Monit In Press; DOI: 10.12659/MSM.952999
Clinical Research
Nasal Mucociliary Clearance and Its Relationship With Disease Severity in Patients With Multiple SclerosisMed Sci Monit In Press; DOI: 10.12659/MSM.952850
Clinical Research
Modified Thoracoabdominal Nerves Block Through the Perichondrial Approach vs Subcostal Transversus Abdomini...Med Sci Monit In Press; DOI: 10.12659/MSM.953976
Most Viewed Current Articles
17 Jan 2024 : Review article 14,176,570
Vaccination Guidelines for Pregnant Women: Addressing COVID-19 and the Omicron VariantDOI :10.12659/MSM.942799
Med Sci Monit 2024; 30:e942799
13 Nov 2021 : Clinical Research 3,762,188
Acceptance of COVID-19 Vaccination and Its Associated Factors Among Cancer Patients Attending the Oncology ...DOI :10.12659/MSM.932788
Med Sci Monit 2021; 27:e932788
14 Dec 2022 : Clinical Research 2,466,310
Prevalence and Variability of Allergen-Specific Immunoglobulin E in Patients with Elevated Tryptase LevelsDOI :10.12659/MSM.937990
Med Sci Monit 2022; 28:e937990
16 May 2023 : Clinical Research 708,927
Electrophysiological Testing for an Auditory Processing Disorder and Reading Performance in 54 School Stude...DOI :10.12659/MSM.940387
Med Sci Monit 2023; 29:e940387