12 February 2021: Database Analysis
Development and Validation of a 5-Gene Autophagy-Based Prognostic Index in Endometrial Carcinoma
Xiaoyan Chen1ABCDEF, Wei Zhang1BCDE, Haiping Zhu2EF, Feng Lin1AEFG*DOI: 10.12659/MSM.928949
Med Sci Monit 2021; 27:e928949






Abstract
BACKGROUND: Endometrial carcinoma (EC) is the most common gynecological malignancy worldwide, and 15–20% of patients with EC have a rapid relapse within 3 years. This study aims to develop an autophagy-related genes (ARGs) signature to predict the prognosis of EC.
MATERIAL AND METHODS: In our study, differentially expressed ARGs were identified by “edgeR” package in R and pathway enrichment analysis was performed to explore biological functions. Univariate and multivariate Cox regression analyses were employed to build autophagy signature. Gene set enrichment analysis (GSEA), Kaplan-Meier curve analysis, and ROC curve analysis were conducted to compare the differences between the high- and low-risk groups.
RESULTS: A total of 60 differentially expressed ARGs (DEARGs) including 34 upregulated and 26 downregulated DEARGs were identified from the TCGAUCEC dataset, with the adjusted P<0.05 and |Fold Change| >1.5. By using univariate and multivariate Cox regression analyses, ERBB2, PRKAB2, GRID2, NRG3, CDKN2A were identified to construct a prognostic signature with AUC 0.673, 0.719, and 0.791, at 1-, 3- and 5- years, respectively. Patients with EC were divided into low- or high-risk group by median risk score, and GSEA showed that low-risk group was enriched in adjacent cells communication pathways while high-risk group was involved in metabolism and immune pathways. The nomograms could also help to guide personal prognostic prediction and therapeutic strategies in EC.
CONCLUSIONS: Our study not only determine 5 ARGs signature that could predict the prognosis of EC but also provide novel insights into the underlying mechanisms of autophagy.
Keywords: Endometrial Neoplasms, nomograms, ROC Curve, AMP-Activated Protein Kinases, Biomarkers, Tumor, Cyclin-Dependent Kinase Inhibitor p16, Databases, Genetic, Neuregulins, Receptor, erbB-2, Receptors, Glutamate
Background
Endometrial cancer (EC) is the most common gynecological malignancy worldwide, with an estimate of 61 880 new cases and 12 160 deaths in 2019 in the United States [1]. Surgery is currently the primary treatment for early-stage EC, while adjuvant radiotherapy or chemotherapy are used in patients with high-risk factors or at advanced stage. However, although most ECs are diagnosed at the early stage (FIGO I–II), the prognosis of recurrent endometrial cancer or advanced stage disease is poor and 15–20% of patients with EC have a rapid relapse within 3 years [2]. Local recurrence or distant metastasis of EC can be clinically important and lead to worse outcomes [3]. Due to the lack of reliable biomarkers for EC, there is an urgent need to explore novel prognostic biomarkers for early risk assessment and high-risk group identification in clinical practice.
Autophagy is a specialized self-degradation mechanism of the catabolic process, which is important in degradation and recycling of cytoplasmic components for maintaining cellular homeostasis [4]. In recent years, growing evidence has suggested that autophagy is critical in the development of various diseases, including cancers [5]. However, the exact role of autophagy in the onset and progression of tumorigenesis remains unknown. Previous studies suggested several autophagy-related genes (ATGs) are involved in the development and progression of various cancers. For example, the upregulated autophagy pathway was reported to be significantly associated with paclitaxel resistance in breast cancer MCF-7 cells [6]. In addition, PI3K/AKT/mTOR and p53 signaling pathways were found to activate or suppress autophagy and may become promising therapeutic targets in endometrial cancer [7]. Although these studies attempted to investigate the role of autophagy in the progression process of EC, most of them paid close attention to an individual gene or a single pathway, without defining an autophagy-based gene signature. In addition, accumulating evidence shows that autophagy-related genes (ATGs) signatures can be used as biomarkers to robustly predict overall survival in several types of tumors, such as gastric cancer, glioblastoma, lung cancer, cutaneous melanoma, and pancreatic ductal adenocarcinoma [8–12]. Therefore, comprehensive analysis of ATGs associated with prognosis are important in understanding the pathogenesis mechanism and potential prognostic implications, as well as clinical diagnosis and treatment in EC.
In this study, we downloaded RNA profiling and clinical data from The Cancer Genome Atlas (TCGA) database, and constructed a ATGs-based signature to predict clinical outcome in EC. Firstly, differentially expressed autophagy-related genes (DEARG) were screened. Subsequently, GO and KEGG analyses were performed to display the enriched pathways of these DEARGs. Univariate and multivariate Cox regression analyses were employed to establish a robust signature associated with prognosis, and were confirmed in a validation dataset. GSEA was conducted to reveal and compare the biological pathways and gene sets between the low- and high-risk groups, which provides more interpretable results to biologists. Finally, a prognostic nomogram was constructed using risk scores and clinicopathologic factors to predict individual survival outcomes.
Material and Methods
DATA ACQUISITION AND PROCESSING:
The Uterine Corpus Endometrial Cancer (UCEC) level 3 RNA-Seq data (HTSeq counts) and corresponding clinicopathological data were obtained from The Cancer Genome Atlas (TGGA) (
DIFFERENTIAL EXPRESSION AND PATHWAY ANALYSIS FOR ARGS:
Differentially expressed genes (DEGs) between tumor and adjacent normal tissues was found using “edgeR” package in R software [13], with the thresholds of adjusted P<0.05 and fold change (FC) >1.5. The intersection of the DEGs and ARGs was considered as the set of differentially expressed autophagy-related genes (DEARGs) for analysis. In addition, volcano plots and heatmap plots were constructed to screen the DEARGs among the datasets.
PATHWAY AND PROTEIN–PROTEIN INTERACTION (PPI) NETWORK ANALYSES:
Gene ontology (GO) terms of biological processes (BP), cellular components (CC), and molecular functions (MF) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses of the DEARGs were conducted using the Database for Annotation, Visualization and Integrated Discovery [14] (DAVID, http://david.ncifcrf.gov/) to predict the possible function of differentially expressed ARGs, and P values of <0.05 were considered to be statistical significant. In addition, the STRING database [15] (http://string-db.org) was employed to establish a PPI network based on differentially expressed ARGs. Cytoscape software [16] was applied for PPI networks visualization and MCODEs were used for modular analysis when the node score cutoff was 0.5 and K-core was 2.
ESTABLISHMENT OF PROGNOSTIC MODEL AND PERFORMANCE ASSESSMENT:
The TCGA-UCEC cohort was randomly partitioned to into a training set and a validation set at a ratio of 1: 1 with the following criteria: (1) samples were randomly divided into training and testing datasets; (2) age distribution, clinical stage, follow-up time, and ratio of death between the 2 sets were similar. Univariate Cox regression analysis was conducted to identify the prognosis-associated DEARGs (
GENE SET ENRICHMENT ANALYSIS (GSEA):
We performed gene set enrichment analysis (GSEA) [17] to identify enrichment of gene sets that differed significantly between the high-risk and low-risk groups (http://www.broadinstitute.org/gsea/index.jsp). The GSEA parameters could be set as follows: 1000 times permutations were performed. The max gene set size was 500 and min size was 15. Enriched biological pathways and co-regulated gene sets with a nominal P value <0.05 were considered to be significant.
NOMOGRAM CONSTRUCTION AND VALIDATION:
A prognostic nomogram was generated by combining autophagy risk scores and other clinicopathologic factors by using the “rms” package (
STATISTICAL ANALYSIS:
All the statistical tests were performed by R software (version 3.5.2,
Results
IDENTIFICATION OF AUTOPHAGY-RELATED RISK SIGNATURE IN UCEC PATIENTS:
The workflow of our study is shown in Figure 1. The expression data of 523 UCEC and 23 adjacent normal tissues were obtained from TCGA. A total of 288 ARGs were obtained from the HADb and MSigDB database. Based on the cutoff criteria of adjusted P<0.05, | FC | >1.5), 60 differentially expressed ARGs (DEARGs) were identified by “edgeR” package in R software. Of these 60 DEARGs, 34 were upregulated and 26 were downregulated. The differentially expressed genes between tumor and adjacent normal tissues are shown in the volcano plot (Figure 2A) and heatmap plot (Figure 2B). In addition, KEGG and GO enrichment analyses were conducted to explore the biological pathways of DEARGs by using DAVID tools. GO analysis revealed that they were significantly enriched in apoptotic process, autophagy, protein binding, and cytoplasm in the aspect of biological process (BP), molecular function (MF), and cellular component (CC) (Figure 3A–3C). KEGG enrichment results suggested that most of them were involved in Pathway in cancer, MicroRNAs in cancer, protein HIF-1 signaling pathway, and ErbB signaling pathway (Figure 3D).
AUTOPHAGY-RELATED PPI NETWORK CONSTRUCTION AND MODULAR ANALYSIS:
A protein–protein interaction (PPI) network of DEARGs was next constructed according to STRING database and visualized by Cytoscape software. A total of 51 nodes and 184 edges mapped from STRING were put into the PPI network (Supplementary Figure 1A). The MCODE plug-in from Cytoscape was employed to search the hub modules in the network with the parameters of Node score cutoff=0.5 and K-core=2. Among them, MCODE1 had 22 nodes and 95 edges, with the highest score (Supplementary Figure 1B), MCODE2 contained 6 nodes and 10 edges (Supplementary Figure 1C), MCODE3 contained 3 nodes and 3 edges (Supplementary Figure 1D), and MCODE4 contained 3 nodes and 3 edges (Supplementary Figure 1E).
CONSTRUCTION OF AUTOPHAGY-RELATED PROGNOSTIC MARKERS IN THE TRAINING COHORT OF TCGA-UCEC:
To identify DEARGs associated with prognosis in patients with EC, univariate Cox proportional hazards regression analysis was performed using the coxph package in R in the training cohort. As a result, only ERBB2, PRKAB2, GRID2, NRG3, CDKN2A were found to be significantly associated with OS in the EC cohort (Table 1). Subsequently, a risk score model was built by multivariate Cox regression based on the 5 genes selected in the univariate Cox regression model, and the risk score for predicting prognosis was obtained as follows:
We applied this model to calculate the risk score for each patient in the training cohort. Using the median risk score as the optimal cutoff, the training cohort was partitioned into high-risk (n=131) and low-risk groups (n=131). In the training cohort, the risk score distribution, overall survival status, and the expression profiles of autophagy genes ranked by the increasing risk score are shown in Figure 4A. Notably, the heatmap displayed that the patients in the high-risk group had significantly higher expression of autophagy genes than those in the low-risk group.
Our data suggested that the patients in the low-risk group tended to have better OS than those in the high-risk group (median time=2.95 years vs 2.46 years, P<0.001, Figure 4B). In addition, to assess the predictive efficacy of the autophagy-related signature, receiver operating characteristic (ROC) curves analysis was conducted. The area under the ROC curves (AUCs) of the signature for 1-, 3-, and 5-year overall survival predictions were 0.673, 0.719, and 0.791, respectively (Figure 4C).
VALIDATION OF AUTOPHAGY-RELATED SIGNATURE FOR PROGNOSTIC PREDICTION:
To confirm that the 5-gene autophagy-related signature had consistent prognostic value in different datasets, we first applied the signature to an independent validation cohort of TCGA-UCEC (n=261). By using the same risk formula, patients were classified into high-risk (n=130) and low-risk (n=131) groups according to the median risk score. Figure 5A shows the distribution of risk score, survival status, and the autophagy genes expression profiles in the validation cohort. Consistent with the training set, the high-risk group had significantly poorer OS than that in the low-risk group (median time=2.20 years vs 2.97 years, log-rank test P<0.001) (Figure 5B). Moreover, the AUCs for 1-, 3-, and 5-year OS predictions for the risk scores were 0.648, 0.736, and 0.713, respectively, which exhibited a good performance to predict survival outcome (Figure 5C).
We additionally validated this autophagy-related signature in the entire TCGA set (n=523). In the entire TCGA set, the distribution pattern of risk score, survival status, and the autophagy genes expression profiles had similar trends to those in the training set (Figure 6A). As shown in Figure 6B, Kaplan-Meier plot analysis indicated that the 5-autophagy gene signature could predict the survival outcomes efficiently, with log-rank test P<0.0001. The 1-, 3-, and 5-year AUCs for the OS were 0.684, 0.708, and 0.743, respectively (Figure 6C). Taken together, these findings suggested that the 5-gene autophagy-related signature could achieve a stable predictive efficacy of the OS in EC.
GENE SET ENRICHMENT ANALYSIS (GSEA) IN CHARACTERIZING HIGH-RISK AND LOW-RISK GROUPS IN EC:
To explore the biological process associated with the autophagy signature, GSEA was conducted to identify signaling pathways that differed between the high-risk and low-risk groups. GSEA results in the entire TCGA set showed that the genes in the low-risk group were enriched in adjacent cells communication pathways, including Axon guidance (NES=1.78, P=0), Cell–cell communication (NES=1.58, P=0.0086), and Tight junction interactions (NES=1.77, P=0.002), while genes in the high-risk group were involved in metabolism and immune pathways, such as Synthesis of Glycosylphosphatidylinositols (GPIs) (NES=−1.85, P=0.01), P53 Hypoxia pathway (NES=−1.84, P=0.004), and IL12 pathway (NES=−1.62, P=0.015) (Figure 7A–7F).
CONSTRUCTION AND VALIDATION OF THE NOMOGRAM IN EC:
To provide clinicians a quantitative prediction of the survival in patients with EC, a prognostic nomogram was constructed by integrating autophagy risk factors and other clinicopathologic features. The nomogram was used to evaluate survival prediction, including the risk scores, age, sex, and TNM stage. As shown in the nomogram (Figure 8A), each variable was assigned points in proportion to its risk contribution to the OS. The C-index to evaluate the OS of the model was 0.785. The calibration curve plot showed a good consistency between actual and ideal observation (Figure 8B–8D).
Discussion
Endometrial carcinoma (EC) remains one of the most common types of gynecological malignancy worldwide, with high mortality rates. The tumor, node, metastasis (TNM) classification is considered as the most clinically useful cancer staging system of EC [18]. However, patients at the same TNM stage can have heterogeneity in clinical outcomes and different response to therapy. Therefore, it is urgent to identify reliable and early indicators that can predict the prognosis of EC. Autophagy is regarded as a highly conserved protective process that plays fundamental roles in cellular stress response and homeostasis maintenance. A variety of autophagy-related genes (ATG) have been found to influence the cellular homeostasis and self-renewal processes. Recent studies have demonstrated that autophagy plays a role as a tumor suppressor at an early stage, but acts as a tumor promoter later on. However, most of these studies were only performed at the individual gene level, and multi-gene patterns were not systematically developed. The present study is the first to comprehensively assess the prognostic value of ARGs signatures of patients with EC.
In this present study, EC patients were obtained from TCGA database and a systematic analysis was conducted to investigate the associations between autophagy signature and clinical outcomes. A total of 60 DEARGs were identified from TCGA-UCEC dataset, including 34 upregulated and 26 downregulated DEARGs. GO and KEGG functional enrichment analyses revealed that the most significantly enriched terms were related to the autophagy process in cancer. Results from KEGG pathways showed that pathway in cancer, MicroRNA pathway in cancer, HIF-1 signaling pathway, and ERBB signaling pathway were significantly enriched, suggesting that deregulated autophagy genes influence a variety of biological processes related to cancer. In addition, Cox regression analysis identified 5 hub prognostic genes – ERBB2, PRKAB2, GRID2, NRG3, and CDKN2A – that could be potential prognostic biomarkers and possible targets for treatment of EC. ERBB2 (also known as HER2) is a 185-kDa transmembrane glycoprotein receptor tyrosine kinase (RTK) that plays a crucial role in activating signaling pathways of cell growth, survival, and differentiation. HER2 was observed to be amplified or overexpressed in 15–30% of breast cancer and gastric cancer samples [19]. Notably, it has been reported that HER2 overexpression (by IHC) exists in 17%–80% of endometrial cancer [20]. Moreover, high HER2 expression in EC has been strongly linked to aggressive status and high risk. Another study reported that HER2 could regulate autophagy through signaling and receptor complexes with autophagy-related genes to affect cancer cell survival and death [21]. For example, HER2 plays crucial roles in modulating autophagic retinal pigment epithelium cell death during oxidative stress [22]. Thus, targeted HER2 therapy or combination therapy might regulate the autophagy process and may be as a promising individualized strategy against EC.
PRKAB2 is the b2 subunit of AMP-activated protein kinase, which maintains cellular energy homeostasis [23]. AMPK can activate energy generating pathways of glycolysis, b-oxidation of fatty acids, and oxidative phosphorylation, and also suppresses energy consuming processes, including lipid synthesis, carbohydrate, and protein translation. PRKAB2 is amplified in many types of cancer [24]. A pan-cancer study demonstrated that PRKAB2 amplification was significantly associated with MYC amplification across various types of cancer, including melanoma, breast, and bladder cancers. In addition, AMPK signaling is important for cellular metabolism, and it mediates autophagic degradation activity and adapts efficiently to the energetic status of the cell [25].
GRID2 (Glutamate Ionotropic Receptor Delta Type Subunit 2) belongs to the ionotropic glutamate receptor family, which was originally described to mediate excitatory neurotransmitters [26]. Functional experiments suggested that changes in the expression level of ionotropic glutamate receptors or their single subunits were critical in proliferation and invasion of cancer cells. A study conducted by Roy et al also showed that GRID2 was a common fragile site easily deleted in some non-primary effusion lymphoma (PEL) tumor cell lines [27]. Similarly,
NRG3 belongs to a growth factor family of an EGF-like domain that binds to ErbB4 or ErbB4/ErbB2 heterodimers exclusively, which stimulates tyrosine phosphorylation of RTK receptor, ultimately contributing to a poor signaling molecule in cancer cells [30,31]. Moreover, NRG3 has reported to bind exclusively to ErbB4 or ErbB4/ErbB2 heterodimers [32], and can affect autophagy through ERBB family signaling.
In general, CDKN2A (p16Ink4a) is considered to be a tumor suppressor, which is often viewed as loss or downregulation in the tumor to negatively regulate G1/S cell cycle progression. In contrast, overexpression of p16Ink4a has also been observed in several kinds of cancers, including invasive endometrial cancers, breast carcinoma, and basal cell carcinoma [33]. Moreover, overexpression of p16Ink4a has been shown to be significantly associated with unfavorable prognosis. In addition to the cell cycle regulation, p16Ink4a protein is also involved in other abnormal signal transduction, such as apoptosis, cell invasion, and angiogenesis, and the activities of these pathways are associated with the overexpression p16Ink4a in cancer [34]. A previous study showed that the cytotoxic effect of the CDK inhibitors enhanced apoptosis in CDKN2A-defective SqCLC cells, which is a sign of autophagy during this process [35].
Although previous studies have shown that these genes are related to the autophagy process, most of them only focused on individual genes. Our study suggests that these genes may influence the cancer development process within a specific pattern, from which autophagy shows a combined effect on the prognosis of EC. Our robust autophagy signature was built by multivariate Cox regression in a training cohort, and validated in a testing cohort and the entire cohort from TCGA, which showed a consistent effect in predictive efficiency. Moreover, the 5-gene autophagy-related signature could successfully divide patients into high-risk and low-risk groups. It was observed that patients with higher risk scores had shortened survival outcomes. Hence, the 5-gene signature could serve as a joint prognostic biomarker in EC and provide further insight into the clinical practice. GSEA results showed that the low-risk group tended to be enriched in adjacent cells communication pathways, while the high-risk group was involved in metabolism and immune pathways, which suggests there are significant differences between the 2 subgroups at pathway levels. Moreover, a nomogram was constructed to predict each individual survival outcome by risk scores and clinicopathologic factors, which could provide a more accurate risk assessment of OS for the patients with EC. Currently, genomic-based classification can stratify endometrial carcinoma into 2 groups: type I carcinomas, which are associated with favorable prognosis, often harbor CTNNB1 mutations and microsatellite instability (MSI), while type II carcinomas are defined by the risk factors of TP53 mutations and high Ki-67 score, which are related to adverse prognosis [36]. We next analyzed the current risk assessing tools of endometrial carcinoma such as POLE, MSI, CTNNB1, and TP53 to compare with the high- or low-risk groups divided by our autophagy gene signature. We observed that MSI (
Taken together, our study reveals that the autophagy-associated pattern could help to distinguish between high-risk and low-risk patients with EC. However, several limitations to our study should be considered. First, our research was only based on RNA-Seq data, and protein expression of these autophagy genes should be validated in the future. Second, other types of clinicopathologic factors such as tumor size and lymph node metastasis, which might be responsible for the prognosis, should be considered. Third, the underlying mechanism of these autophagy genes remains unclear, and in vitro or in vivo laboratory experiments are needed to confirm our findings.
Conclusions
In summary, this present study developed a robust 5-gene autophagy-related signature that can accurately predict OS outcomes in patients with EC. We hope this novel autophagy-based prognostic signature will be helpful for the clinical guidance of individualized treatment of patients with EC.
Figures
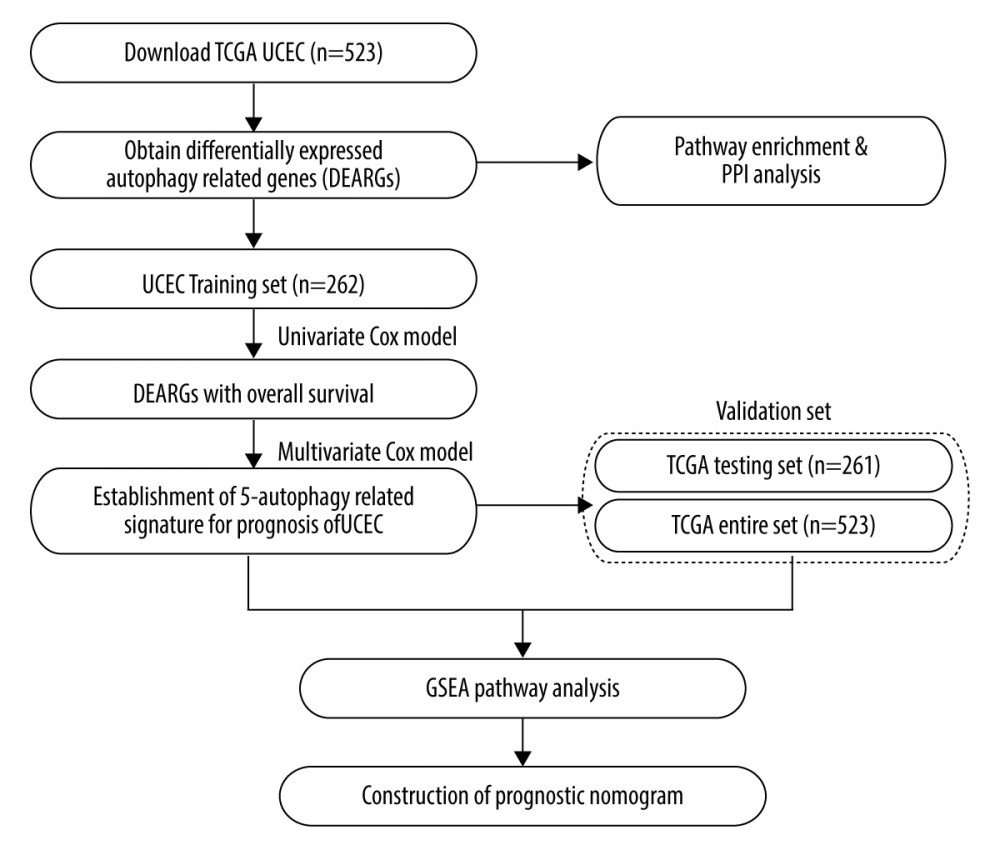 Figure 1. An entire workflow for identification of UCEC prognostic autophagy signature in our study.
Figure 1. An entire workflow for identification of UCEC prognostic autophagy signature in our study. 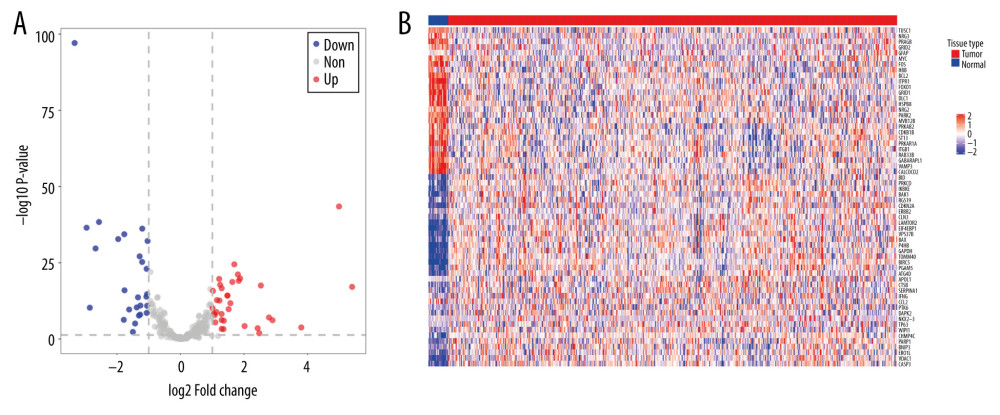 Figure 2. The expression of differential autophagy-related genes (ARGs) in TCGA-UCEC cohort. The expression of differential ARGs between EC tissues (n=523) and normal tissues (n=23) was shown in the plots. (A) In the volcano plot, red dots stand for upregulated ARGs, blue dots stand for downregulated ARGs, and the gray dots stand for the ARGs that were not differentially expressed. (B) The heatmap plot shows ARGs between EC tissues and normal tissues. Red color is upregulated ARGs and blue color is downregulated ARGs.
Figure 2. The expression of differential autophagy-related genes (ARGs) in TCGA-UCEC cohort. The expression of differential ARGs between EC tissues (n=523) and normal tissues (n=23) was shown in the plots. (A) In the volcano plot, red dots stand for upregulated ARGs, blue dots stand for downregulated ARGs, and the gray dots stand for the ARGs that were not differentially expressed. (B) The heatmap plot shows ARGs between EC tissues and normal tissues. Red color is upregulated ARGs and blue color is downregulated ARGs. 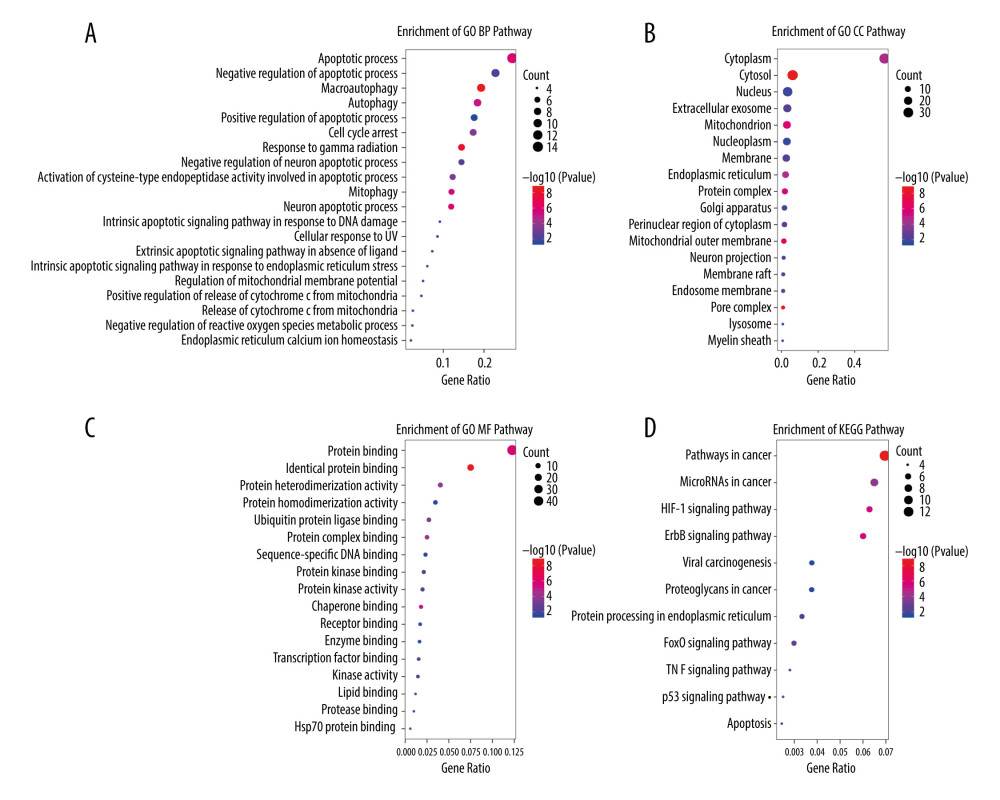 Figure 3. KEGG and GO functional enrichment analysis for differentially expressed autophagy-related genes. The vertical axis represents GO or KEGG pathways. The horizontal axis represents the number of genes assigned to the corresponding annotation (A) The top 20 significant terms of CC. (B) The top 20 significant terms of BP. (C) The top 20 significant terms of MF. (D) The top 11 significant terms of KEGG pathways. BP – biological process; CC – cellular component; MF – molecular function.
Figure 3. KEGG and GO functional enrichment analysis for differentially expressed autophagy-related genes. The vertical axis represents GO or KEGG pathways. The horizontal axis represents the number of genes assigned to the corresponding annotation (A) The top 20 significant terms of CC. (B) The top 20 significant terms of BP. (C) The top 20 significant terms of MF. (D) The top 11 significant terms of KEGG pathways. BP – biological process; CC – cellular component; MF – molecular function. 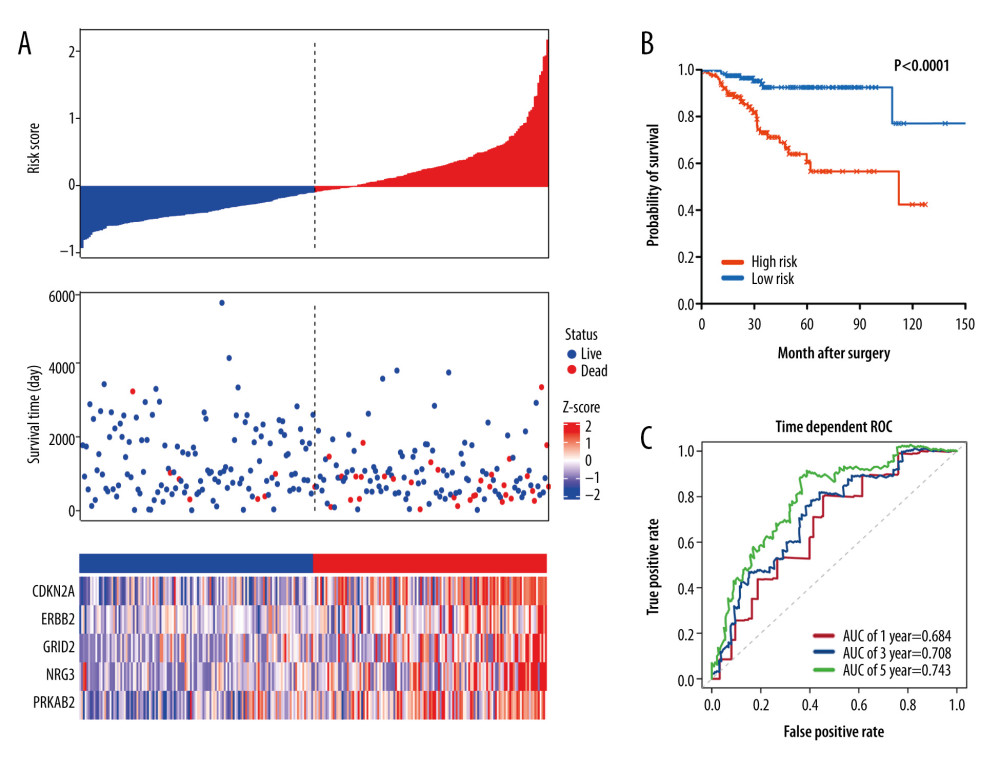 Figure 4. Characteristics of differential expression autophagy-related genes (DEARGs) autophagy signature, Kaplan-Meier plot, and AUC curves in the TCGA training set. (A) Distribution of risk score, patient survival status, the heatmap of autophagy-related gene expression profiles. (B) Kaplan-Meier curves of the prognostic predictors for high-risk and low-risk patients with UCEC. (C) Time-dependent ROC curves for the efficacy evaluation of the 5-gene autophagy-related signature.
Figure 4. Characteristics of differential expression autophagy-related genes (DEARGs) autophagy signature, Kaplan-Meier plot, and AUC curves in the TCGA training set. (A) Distribution of risk score, patient survival status, the heatmap of autophagy-related gene expression profiles. (B) Kaplan-Meier curves of the prognostic predictors for high-risk and low-risk patients with UCEC. (C) Time-dependent ROC curves for the efficacy evaluation of the 5-gene autophagy-related signature. 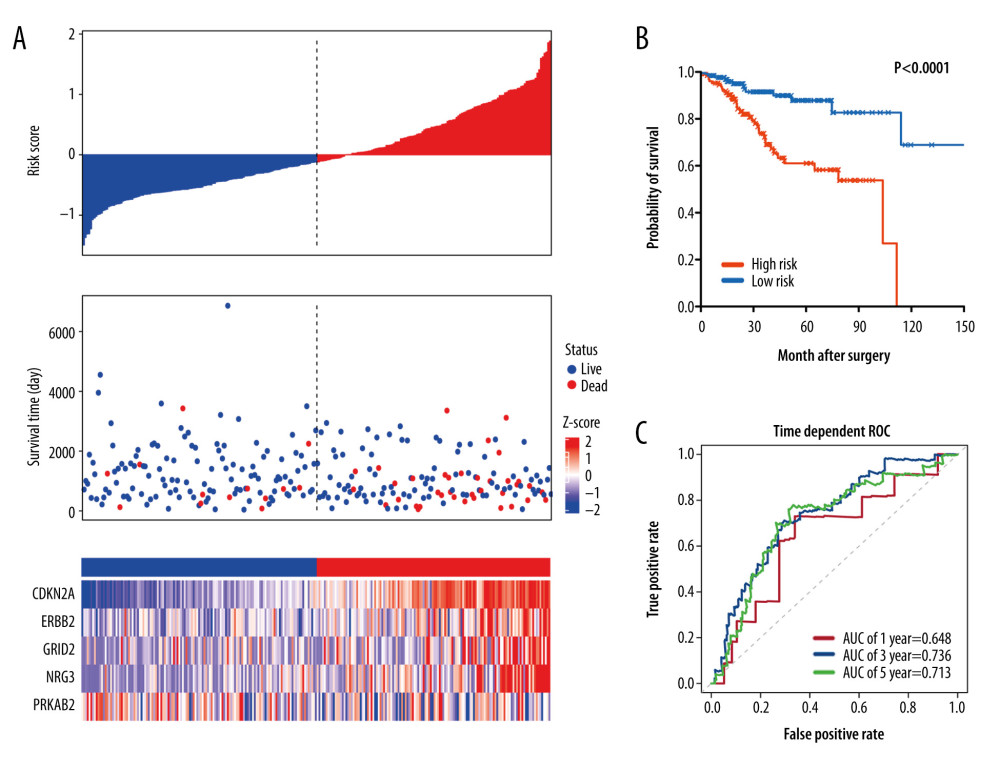 Figure 5. Characteristics of differential expression autophagy-related genes (DEARGs) autophagy signature, Kaplan-Meier plot, and AUC curves in the TCGA testing set. (A) Distribution of risk score, patient survival status, the heatmap of autophagy-related gene expression profiles. (B) Kaplan-Meier curves of the prognostic predictors for high-risk and low-risk patients with UCEC. (C) Time-dependent ROC curves for the efficacy evaluation of the 5-gene autophagy-related signature.
Figure 5. Characteristics of differential expression autophagy-related genes (DEARGs) autophagy signature, Kaplan-Meier plot, and AUC curves in the TCGA testing set. (A) Distribution of risk score, patient survival status, the heatmap of autophagy-related gene expression profiles. (B) Kaplan-Meier curves of the prognostic predictors for high-risk and low-risk patients with UCEC. (C) Time-dependent ROC curves for the efficacy evaluation of the 5-gene autophagy-related signature. 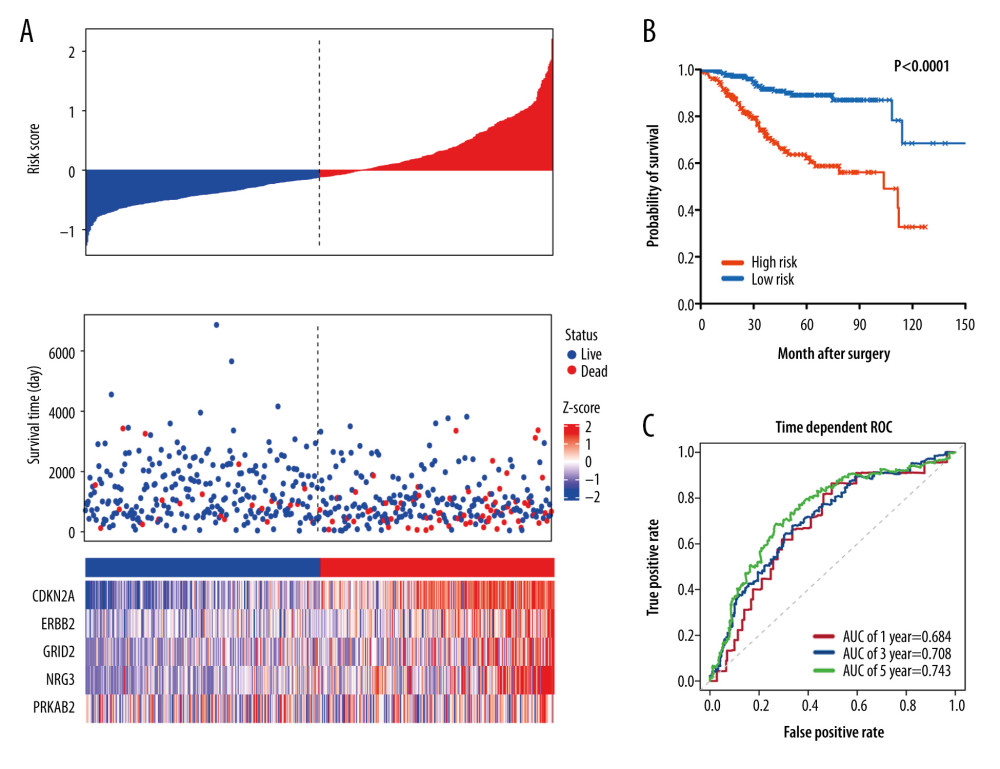 Figure 6. Characteristics of differential expression autophagy-related genes (DEARGs) autophagy signature, Kaplan-Meier plot, and AUC curves in the TCGA entire set. (A) Distribution of risk score, patient survival status, and heatmap of autophagy-related gene expression profiles. (B) Kaplan-Meier curves of the prognostic predictors for high-risk and low-risk patients with UCEC. (C) Time-dependent ROC curves for the efficacy evaluation of the 5-gene autophagy-related signature.
Figure 6. Characteristics of differential expression autophagy-related genes (DEARGs) autophagy signature, Kaplan-Meier plot, and AUC curves in the TCGA entire set. (A) Distribution of risk score, patient survival status, and heatmap of autophagy-related gene expression profiles. (B) Kaplan-Meier curves of the prognostic predictors for high-risk and low-risk patients with UCEC. (C) Time-dependent ROC curves for the efficacy evaluation of the 5-gene autophagy-related signature. 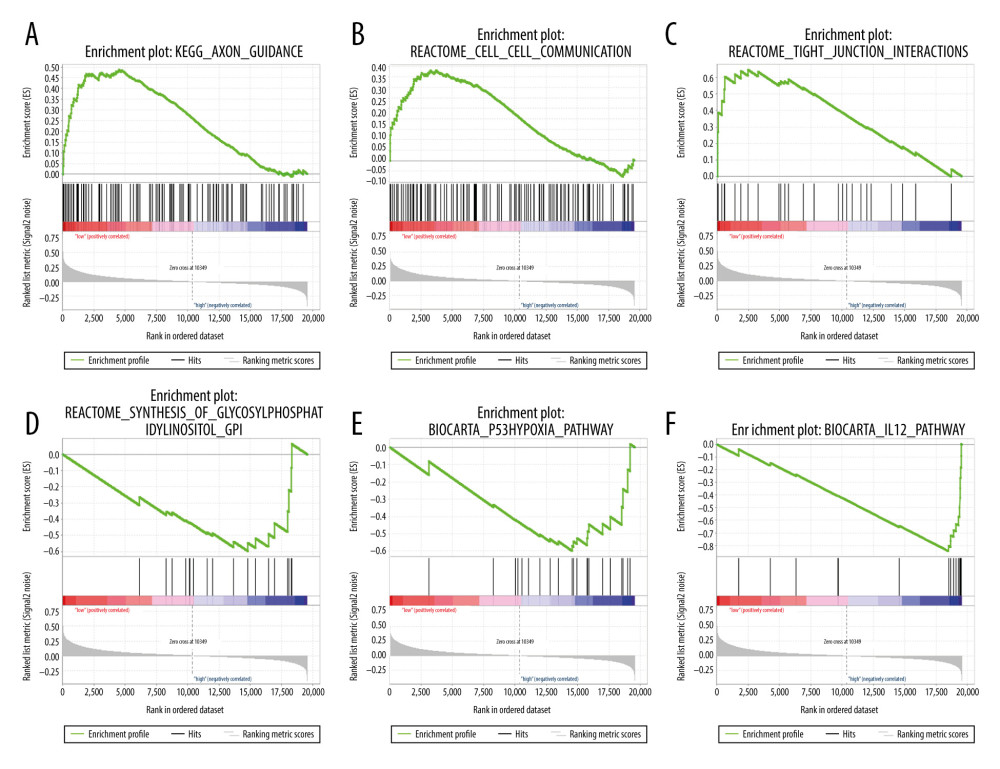 Figure 7. GSEA enrichment analysis in TCGA-UCEC. (A) “Axon guidance”, (B) “Cell-cell communication”, (C) “Tight junction interactions”, (D) “Synthesis of Glycosylphosphatidylinositols (GPIs)”, (E) “P53 Hypoxia pathway”, (F) “IL12 pathway”.
Figure 7. GSEA enrichment analysis in TCGA-UCEC. (A) “Axon guidance”, (B) “Cell-cell communication”, (C) “Tight junction interactions”, (D) “Synthesis of Glycosylphosphatidylinositols (GPIs)”, (E) “P53 Hypoxia pathway”, (F) “IL12 pathway”. 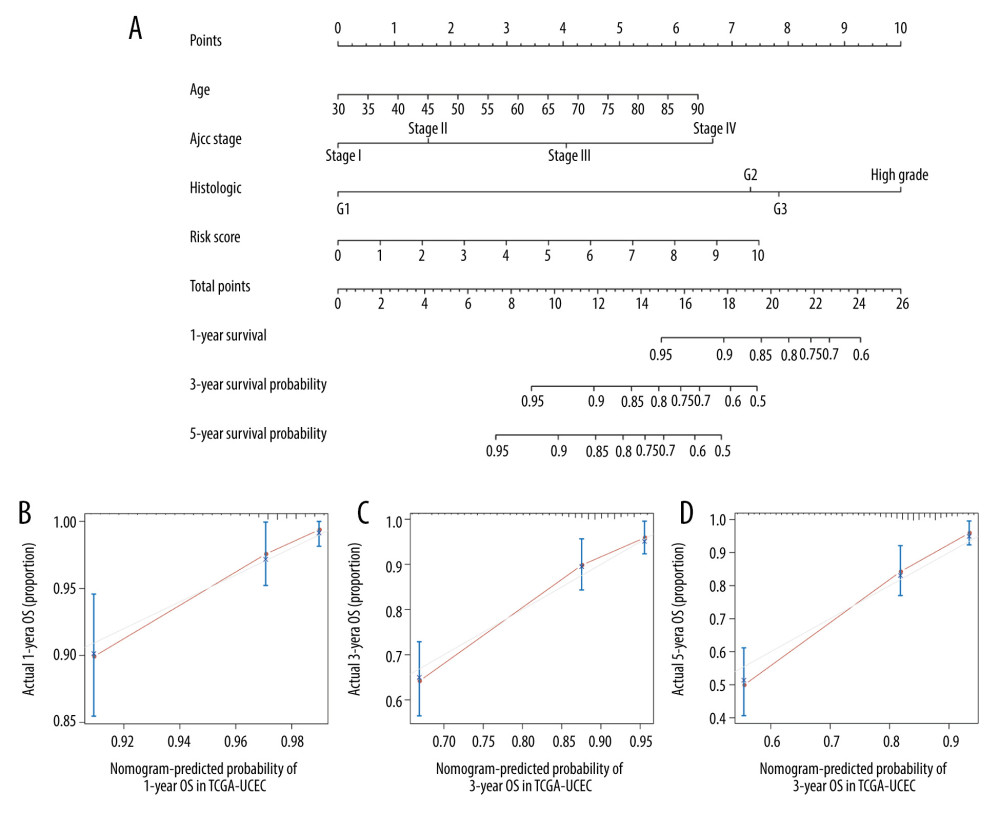 Figure 8. Nomogram for predicting the 1-, 3-, and 5-year survival with risk score. (A) A prognostic nomogram for patients with UCEC in TCGA; (B–D) Calibration curves for the nomogram at 1, 3, and 5 years.
Figure 8. Nomogram for predicting the 1-, 3-, and 5-year survival with risk score. (A) A prognostic nomogram for patients with UCEC in TCGA; (B–D) Calibration curves for the nomogram at 1, 3, and 5 years. 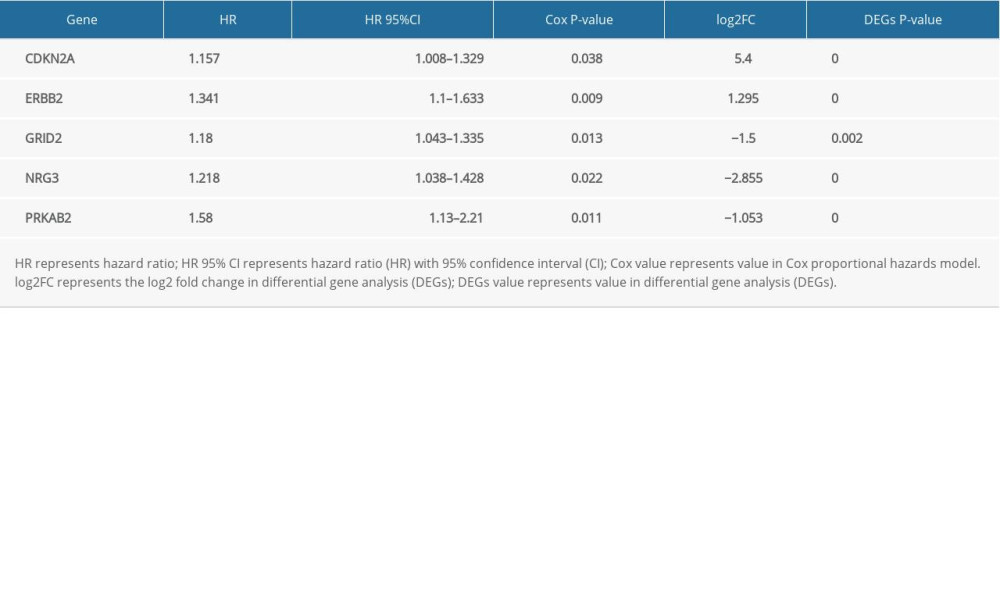
References
1. Siegel RL, Miller KD, Jemal A, Cancer statistics, 2019: Cancer J Clin, 2019; 69(1); 7-34
2. Sohaib SA, Houghton SL, Meroni R, Recurrent endometrial cancer: Patterns of recurrent disease and assessment of prognosis: Clin Radiol, 2007; 62(1); 28-34 discussion 35–36
3. Kurra V, Krajewski KM, Jagannathan J, Typical and atypical metastatic sites of recurrent endometrial carcinoma: Cancer Imaging, 2013; 13; 113-22
4. Das G, Shravage BV, Baehrecke EH, Regulation and function of autophagy during cell survival and cell death: Cold Spring Harb Perspect Biol, 2012; 4(6); e008813
5. Yun CW, Lee SH, The roles of autophagy in cancer: Int J Mol Sci, 2018; 19(11); 3466
6. Ajabnoor GM, Crook T, Coley HM, Paclitaxel resistance is associated with switch from apoptotic to autophagic cell death in MCF-7 breast cancer cells: Cell Death Dis, 2012; 3; e260
7. Nunez-Olvera SI, Gallardo-Rincon D, Puente-Rivera J, Autophagy machinery as a promising therapeutic target in endometrial cancer: Front Oncol, 2019; 9; 1326
8. Yue P, Zhu C, Gao Y, Development of an autophagy-related signature in pancreatic adenocarcinoma: Biomed Pharmacother, 2020; 126; 110080
9. Qiu J, Sun M, Wang Y, Chen B, Identification and validation of an individualized autophagy-clinical prognostic index in gastric cancer patients: Cancer Cell Int, 2020; 20; 178
10. Wang QW, Liu HJ, Zhao Z, Prognostic correlation of autophagy-related gene expression-based risk signature in patients with glioblastoma: Onco Targets Ther, 2020; 13; 95-107
11. Liu Y, Wu L, Ao H, Prognostic implications of autophagy-associated gene signatures in non-small cell lung cancer: Aging (Albany NY), 2019; 11(23); 11440-62
12. Tang DY, Ellis RA, Lovat PE, Prognostic impact of autophagy biomarkers for cutaneous melanoma: Front Oncol, 2016; 6; 236
13. Robinson MD, McCarthy DJ, Smyth GK, edgeR: A bioconductor package for differential expression analysis of digital gene expression data: Bioinformatics, 2010; 26(1); 139-40
14. Huang DW, Sherman BT, Lempicki RA, Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources: Nat Protoc, 2009; 4(1); 44-57
15. Szklarczyk D, Gable AL, Lyon D, STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets: Nucleic Acids Res, 2019; 47(D1); D607-13
16. Shannon P, Markiel A, Ozier O, Cytoscape: A software environment for integrated models of biomolecular interaction networks: Genome Res, 2003; 13(11); 2498-504
17. Subramanian A, Tamayo P, Mootha VK, Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles: Proc Natl Acad Sci USA, 2005; 102(43); 15545-50
18. Sher YP, Shih JY, Yang PC, Prognosis of non-small cell lung cancer patients by detecting circulating cancer cells in the peripheral blood with multiple marker genes: Clin Cancer Res, 2005; 11(1); 173-79
19. Iqbal N, Iqbal N, Human epidermal growth factor receptor 2 (HER2) in cancers: Overexpression and therapeutic implications: Mol Biol Int, 2014; 2014 852748
20. Diver EJ, Foster R, Rueda BR, Growdon WB, The therapeutic challenge of targeting HER2 in endometrial cancer: Oncologist, 2015; 20(9); 1058-68
21. Henson E, Chen Y, Gibson S, EGFR family members’ regulation of autophagy is at a crossroads of cell survival and death in cancer: Cancers (Basel), 2017; 9(4); 27
22. Sheu SJ, Chen JL, Bee YS, ERBB2-modulated ATG4B and autophagic cell death in human ARPE19 during oxidative stress: PLoS One, 2019; 14(3); e0213932
23. Marin-Aguilar F, Pavillard LE, Giampieri F, Adenosine monophosphate (AMP)-activated protein kinase. A new target for nutraceutical compounds: Int J Mol Sci, 2017; 18(2); 288
24. Ross FA, MacKintosh C, Hardie DG, AMP-activated protein kinase: A cellular energy sensor that comes in 12 flavours: FEBS J, 2016; 283(16); 2987-3001
25. Tamargo-Gomez I, Marino G, AMPK: Regulation of metabolic dynamics in the context of autophagy: Int J Mol Sci, 2018; 19(12); 3812
26. Stepulak A, Rola R, Polberg K, Ikonomidou C, Glutamate and its receptors in cancer: J Neural Transm (Vienna), 2014; 121(8); 933-44
27. Roy D, Sin SH, Damania B, Dittmer DP, Tumor suppressor genes FHIT and WWOX are deleted in primary effusion lymphoma (PEL) cell lines: Blood, 2011; 118(7); e32-39
28. Hazan I, Hofmann TG, Aqeilan RI, Tumor suppressor genes within common fragile sites are active players in the DNA damage response: PLoS Genet, 2016; 12(12); e1006436
29. Selimi F, Lohof AM, Heitz S, Lurcher GRID2-induced death and depolarization can be dissociated in cerebellar Purkinje cells: Neuron, 2003; 37(5); 813-19
30. Hijazi MM, Young PE, Dougherty MK, NRG-3 in human breast cancers: Activation of multiple erbB family proteins: Int J Oncol, 1998; 13(5); 1061-67
31. Hobbs SS, Coffing SL, Le AT, Neuregulin isoforms exhibit distinct patterns of ErbB family receptor activation: Oncogene, 2002; 21(55); 8442-52
32. Lebrasseur NK, Cote GM, Miller TA, Regulation of neuregulin/ErbB signaling by contractile activity in skeletal muscle: Am J Physiol Cell Physiol, 2003; 284(5); C1149-55
33. Milde-Langosch K, Bamberger AM, Rieck G, Overexpression of the p16 cell cycle inhibitor in breast cancer is associated with a more malignant phenotype: Breast Cancer Res Treat, 2001; 67(1); 61-70
34. Romagosa C, Simonetti S, Lopez-Vicente L, p16(Ink4a) overexpression in cancer: A tumor suppressor gene associated with senescence and high-grade tumors: Oncogene, 2011; 30(18); 2087-97
35. Jeong EH, Lee TG, Ko YJ, Anti-tumor effect of CDK inhibitors on CDKN2A-defective squamous cell lung cancer cells: Cell Oncol (Dordr), 2018; 41(6); 663-75
36. Kandoth C, Schultz N, XCherniack ADCancer Genome Atlas Research Network, Integrated genomic characterization of endometrial carcinoma: Nature, 2013; 497(7447); 67-73
Figures
Figure 1. An entire workflow for identification of UCEC prognostic autophagy signature in our study.Figure 2. The expression of differential autophagy-related genes (ARGs) in TCGA-UCEC cohort. The expression of differential ARGs between EC tissues (n=523) and normal tissues (n=23) was shown in the plots. (A) In the volcano plot, red dots stand for upregulated ARGs, blue dots stand for downregulated ARGs, and the gray dots stand for the ARGs that were not differentially expressed. (B) The heatmap plot shows ARGs between EC tissues and normal tissues. Red color is upregulated ARGs and blue color is downregulated ARGs.Figure 3. KEGG and GO functional enrichment analysis for differentially expressed autophagy-related genes. The vertical axis represents GO or KEGG pathways. The horizontal axis represents the number of genes assigned to the corresponding annotation (A) The top 20 significant terms of CC. (B) The top 20 significant terms of BP. (C) The top 20 significant terms of MF. (D) The top 11 significant terms of KEGG pathways. BP – biological process; CC – cellular component; MF – molecular function.Figure 4. Characteristics of differential expression autophagy-related genes (DEARGs) autophagy signature, Kaplan-Meier plot, and AUC curves in the TCGA training set. (A) Distribution of risk score, patient survival status, the heatmap of autophagy-related gene expression profiles. (B) Kaplan-Meier curves of the prognostic predictors for high-risk and low-risk patients with UCEC. (C) Time-dependent ROC curves for the efficacy evaluation of the 5-gene autophagy-related signature.Figure 5. Characteristics of differential expression autophagy-related genes (DEARGs) autophagy signature, Kaplan-Meier plot, and AUC curves in the TCGA testing set. (A) Distribution of risk score, patient survival status, the heatmap of autophagy-related gene expression profiles. (B) Kaplan-Meier curves of the prognostic predictors for high-risk and low-risk patients with UCEC. (C) Time-dependent ROC curves for the efficacy evaluation of the 5-gene autophagy-related signature.Figure 6. Characteristics of differential expression autophagy-related genes (DEARGs) autophagy signature, Kaplan-Meier plot, and AUC curves in the TCGA entire set. (A) Distribution of risk score, patient survival status, and heatmap of autophagy-related gene expression profiles. (B) Kaplan-Meier curves of the prognostic predictors for high-risk and low-risk patients with UCEC. (C) Time-dependent ROC curves for the efficacy evaluation of the 5-gene autophagy-related signature.Figure 7. GSEA enrichment analysis in TCGA-UCEC. (A) “Axon guidance”, (B) “Cell-cell communication”, (C) “Tight junction interactions”, (D) “Synthesis of Glycosylphosphatidylinositols (GPIs)”, (E) “P53 Hypoxia pathway”, (F) “IL12 pathway”.Figure 8. Nomogram for predicting the 1-, 3-, and 5-year survival with risk score. (A) A prognostic nomogram for patients with UCEC in TCGA; (B–D) Calibration curves for the nomogram at 1, 3, and 5 years. In Press
Clinical Research
Effects of Single-Bout Endurance Exercise Intensity on Peripheral Neurotrophic Factors in Patients With Isc...Med Sci Monit In Press; DOI: 10.12659/MSM.952089
Review article
Anisodus tanguticus in Cancer Research: A Review of Traditional Use, Phytochemistry, Extraction Methods, an...Med Sci Monit In Press; DOI: 10.12659/MSM.952999
Clinical Research
Nasal Mucociliary Clearance and Its Relationship With Disease Severity in Patients With Multiple SclerosisMed Sci Monit In Press; DOI: 10.12659/MSM.952850
Clinical Research
Modified Thoracoabdominal Nerves Block Through the Perichondrial Approach vs Subcostal Transversus Abdomini...Med Sci Monit In Press; DOI: 10.12659/MSM.953976
Most Viewed Current Articles
17 Jan 2024 : Review article 14,176,570
Vaccination Guidelines for Pregnant Women: Addressing COVID-19 and the Omicron VariantDOI :10.12659/MSM.942799
Med Sci Monit 2024; 30:e942799
13 Nov 2021 : Clinical Research 3,762,188
Acceptance of COVID-19 Vaccination and Its Associated Factors Among Cancer Patients Attending the Oncology ...DOI :10.12659/MSM.932788
Med Sci Monit 2021; 27:e932788
14 Dec 2022 : Clinical Research 2,466,310
Prevalence and Variability of Allergen-Specific Immunoglobulin E in Patients with Elevated Tryptase LevelsDOI :10.12659/MSM.937990
Med Sci Monit 2022; 28:e937990
16 May 2023 : Clinical Research 708,927
Electrophysiological Testing for an Auditory Processing Disorder and Reading Performance in 54 School Stude...DOI :10.12659/MSM.940387
Med Sci Monit 2023; 29:e940387