11 July 2020: Database Analysis
Clinicopathological Characteristics and Survival Outcomes of Primary Hepatic Neuroendocrine Tumor: A Surveillance, Epidemiology, and End Results (SEER) Population-Based Study
Yu-feng Li12ABCE, Qiu-qiang Zhang12BF, Wei-lin Wang1234AEG*DOI: 10.12659/MSM.923375
Med Sci Monit 2020; 26:e923375






Abstract
BACKGROUND: Primary hepatic neuroendocrine tumor (PHNET) is a rare primary liver tumor that remains poorly understood. Here, we explored the clinicopathological characteristics and survival outcomes of PHNET patients.
MATERIAL AND METHODS: PHNET patients diagnosed between 1988 and 2015 in the Surveillance, Epidemiology, and End Results (SEER) database were enrolled in the cohort. Kaplan-Meier analysis was used to determine the survival outcomes. Multivariable Cox regression models were used to identify the risk factors for overall survival (OS) and disease-specific survival (DSS).
RESULTS: A total of 291 PHNET patients from the SEER database met the inclusion criteria for analysis. The majority of the patients were female (53.6%), white (77.7%), and married (49.5%). The 1-, 3-, and 5-year OS were 57.1%, 39.4%, and 30.2%, and the 1-, 3-, and 5-year DSS rates were 61.3%, 44.3%, and 36.7%, respectively. Multivariate Cox regression models showed that older age, unmarried status, poor differentiated grade, and no tumor-directed surgery were independent risk factors for poor OS and DSS.
CONCLUSIONS: Older age, unmarried status, poor differentiated grade, and no tumor-directed surgery were associated with poorer prognosis of PHNET. Surgical resection is an effective and reliable treatment method for patients with PHNET.
Keywords: Epidemiology, Liver Neoplasms, neuroendocrine tumors, Pathology, Clinical, Aged, 80 and over, Carcinoma, Neuroendocrine, Intestinal Neoplasms, Pancreatic Neoplasms, Proportional Hazards Models, Risk Factors, SEER Program, Survival Rate
Background
Neuroendocrine tumors (NETs) are a group of heterogeneous neoplasms that derive from the neuroendocrine system. The incidence of NETs has been increasing in the past 30 years [1,2]. NETs mainly occur in the gastrointestinal system, with 30.8% in the small intestines, 26.3% in the rectum, 17.6% in the colon, 12.1% in the pancreas, and 5.7% in the appendix [3]. The liver is considered as the most common metastatic site of NETs [4]. However, primary hepatic neuroendocrine tumors (PHNET), by contrast, are extremely rare and only comprise approximately 0.3% of all NETs [4]. Since the first case of PHNET was reported by Edmonson 62 years ago [5], the number of all reported PHNET cases is less than 200 in the English literature [4,6–21]. Diagnosis of PHNET relies on pathological analysis and the exclusion of metastasized NETs. Surgical resection is considered as the mainstay for treatment of this disease [4].
Due to the rarity of PHNET, little is known about it. The aim of this study was to explore the clinicopathological characteristics and survival outcomes of PHNET patients, diagnosed from 1988 to 2015, in the Surveillance, Epidemiology, and End Results (SEER) database.
Material and Methods
DATA SOURCE AND STUDY POPULATION:
Our study used data from the National Cancer Institute’s 18-registry SEER research database (1973–2015), based on the November 2017 submission, which represented approximately 30% of the US population. We identified PHNET patients diagnosed between 1988 and 2015 using SEER*Stat software (version 8.3.6). Patients were retrieved according to the International Classification of Disease for Oncology (3rd edition) (ICD-O-3) site record for the liver (C22.0). The following ICD-O-3 histology codes were involved: 8013, 8041, 8240, 8241, 8242, 8243, 8244, 8245, 8246, and 8249. The study enrollment criterion was patients who had a positive histological diagnosis with PHNET as a primary disease. Patients were excluded if they had incomplete follow-up information or their survival month was recorded as 0. Informed consent was not required as our study did not use any identifiable human subjects or information.
Patient characteristics retrieved from the database included age at diagnosis, sex, race, marital status, histological grade, SEER stage, and surgery information. Patients were divided according to age at diagnosis into 2 groups: ≤65 years and >65 years. Race was divided into white, black, others (including American Indian/Alaska Native, Asian/Pacific Islander, and unknown). Marital status was categorized as married and unmarried (including divorced, widowed, separated, and single). Histological grade was classified according to the degree of the tumor differentiation, including well differentiated, I; moderately differentiated, II; poorly differentiated, III; undifferentiated, IV. SEER stage was classified as localized, regional, and distant. Surgical treatment was categorized into 2 groups: a tumor-directed surgery group and a no tumor-directed surgery group.
The primary outcomes of our study were overall survival (OS) and disease-specific survival (DSS). OS was defined from the date of diagnosis to death due to any cause. DSS was defined from the date of diagnosis to death due to PHNET.
STATISTICAL ANALYSIS:
Survival curves were created using the Kaplan-Meier method, and differences between groups were compared using the log-rank test. Multivariable Cox proportional hazards regression models were used to identify the potential risk factors for prognosis. All P-values were based on a two-sided significance level, and P-values <0.05 were considered statistically significant. All statistical analyses were conducted using GraphPad Prism software 8.0 (GraphPad, San Diego, CA, USA) and SPSS 19 for Windows (SPSS, Inc., Chicago, IL, USA).
Results
A total of 291 PHNET patients were involved in our study cohort. Patient demographics and the clinicopathological characteristics are shown in Table 1. The median age was 63 years (range 18–91), and 127 patients (43.6%) were older than 65 years. As shown in Figure 1, the incidence of PHNET peaked in 50–80 year. There was no significant difference in sex-specific distribution, with 156 females (53.6%) and 135 males (46.4%). A total of 226 patients (77.7%) were white, and 144 patients (49.5%) were married. Histologically, 50 (17.2%) were grade I (G1), 22 (7.5%) were grade II (G2), 50 (17.2%) were grade III (G3), 13 (4.5%) were grade IV (G4), and 156 (53.6%) were unknown. According to SEER stage, 91 patients (31.3%) had localized disease, 67 patients (23.0%) had regional disease, and 63 patient (21.7%) had distant disease. There were 76 patients (26.1%) who underwent tumor-directed surgery, while the remaining 215 patients (73.9%) did not receive surgery.
Then, we performed Kaplan-Meier analysis for evaluating OS and DSS of PHNET (Figure 2). The 1-, 3-, and 5-year OS were 57.1%, 39.4%, and 30.2%, and the 1-, 3-, and 5-year DSS were 61.3%, 44.3%, and 36.7%, respectively. Kaplan-Meier survival analysis with the log-rank test revealed that age, marital status, histological grade, SEER stage, and tumor-directed surgery were significantly associated with OS (Figure 3) and DSS (Figure 4). In brief, patients with younger age, G1/G2 tumors, and localized disease had better survival than those with older age, G3/G4 tumors, and regional/distant disease. Moreover, married patients tended to have longer survival than unmarried patients. We also found that who patients received tumor-directed surgery tended to have better survival outcome in both OS and CSS. Multivariate Cox regression models using proportional hazards ratios were conducted to identify independent risk factors for survival of PHNET (Table 2). The results showed that older age, unmarried status, poor differentiated grade, and no tumor-directed surgery were independent risk factors for poorer OS and DSS.
Discussion
PHNET is an extremely rare primary tumor of liver and only comprise approximately 0.3% of all NETs. Due to the rarity of PHNET, the clinical characteristics and survival outcome of the disease are still not fully understood. In the present study, we first explored the clinicopathological features and survival outcomes of PHNET using detailed data from the SEER database.
A systematic review of 124 PHNET cases showed that the mean age was 51.9±16.5 years and found no clear sex-specific patterns (50.8% female and 49.2% male) [4]. In a Korean retrospective study of 12 PHNET patients, the median age was 66.5 years and 7 patients (58.3%) were male [6]. Chen et al. reported that among 28 patients with PHNET, the mean age was 53 years (range 32–76) with a male-to-female ratio of 1.15: 1 [13]. An another single-center PHNET study of 22 Chinese patients showed that the median age was 49 years (range 37–82), with 12 males and 10 females [14]. In our study, the median age of the 291 PHNET patients was 63 years (range 18–91), and 46.4% of patients were male. These results indicated that PHNET occurred more in the middle-aged and elderly patients and it is not a sex-specific disease.
Previous studies have revealed that clinical tumor markers such as alpha-fetoprotein (AFP), carcinoembryonic antigen (CEA), and cancer-antigen (CA) 199 have no diagnostic value in PHNET [19,22]. Medical imaging examinations such as ultrasound, computed tomography, and magnetic resonance imaging also have low sensitivity and specificity for diagnosis of PHNET [9,23–25]. Therefore, pathological diagnosis based on histological and immunohistochemical evaluation is regarded as the final diagnosis standard for PHNET. Because the liver is regarded as the most common metastatic site of NETs, the diagnosis of PHNET should exclude metastasized NETs. Based on mitotic rates and Ki-67 proliferation index (PI), neuroendocrine tumors are classified into 3 categories in the WHO digestive system classification [26], but in the SEER database, PHNET is divided into 4 histological grades according to the degree of tumor differentiation. As shown in our results, patients with G1/G2 histological grade tended to have better OS and DSS than those with G3/G4, and histological grade was an independent risk predictor of the prognosis of PHNET.
Previous studies have not clearly elucidated the risk factors for prognosis of PHNET. Chen et al. reported that high expression of Ki-67 was an independent prognostic factor for PHNET [13]. Shi et al. found that histological grade of G3 was an independent factor for recurrence-free survival [14]. In the present study, we found that older age, unmarried status, G3/G4 tumors, regional/distant disease, and lack of surgery were significantly associated with poorer OS and DSS. Moreover, older age, unmarried status, poor differentiated grade, and no tumor-directed surgery were independent risk factors for survival of PHNET. These results will help clinicians to assess the survival and prognosis of PHNET patients.
Surgical resection remains the main treatment modality for PHNET. Patients who received surgery had better prognosis. Iwo et al. reported that the 5-year survival rate was 74% in their 53-patient cohort [27]. Knox et al. found a similar 5-year survival rate of 78% in their analysis of 48 patients with PHNET [28]. In the present study, the 5-year OS was 71.9% and CSS was 75.7% in patients who received surgery, compared to the 15.6% and 21.5% in those did not receive surgery. These results indicate that tumor-directed surgery can significantly improve the prognosis of PHNET. However, previous studies have indicated that the recurrence rates after surgery were as high as 20–40% [14,27,28]. Therefore, close follow-up is recommended in the postoperative period for patients of PHNET. Apart from surgery, other treatment modalities of PHNET include transcatheter arterial chemoembolization (TACE) [19,29], radiofrequency ablation (RFA) [30], percutaneous ethanol injection treatment (PEIT) [22], liver transplantation [18,31], somatostatin analogues [32], and chemotherapy. However, the role and effectiveness of these treatment modalities remain unclear and need further study.
Our study has certain limitations. First, the SEER database does not contain important information such as the details of surgery and postoperative complications, as well as the type of chemotherapy and radiotherapy; these influence patient survival but we could not adjust for them in our analysis. Second, this study had a retrospective design, and due to lack of details about clinical features in the SEER data, we could not prevent all bias. Thus, the present results need to be verified in larger studies.
Conclusions
Our study assessed the largest PHNET population based on the SEER database to date. PHNET is a rare primary liver tumor and its prognosis is generally favorable after active treatment. Moreover, our study found that older age, unmarried status, poor differentiated grade, and no tumor-directed surgery were associated with poorer prognosis of PHNET. Surgical resection is the most effective and reliable treatment method for patients with PHNET.
Figures
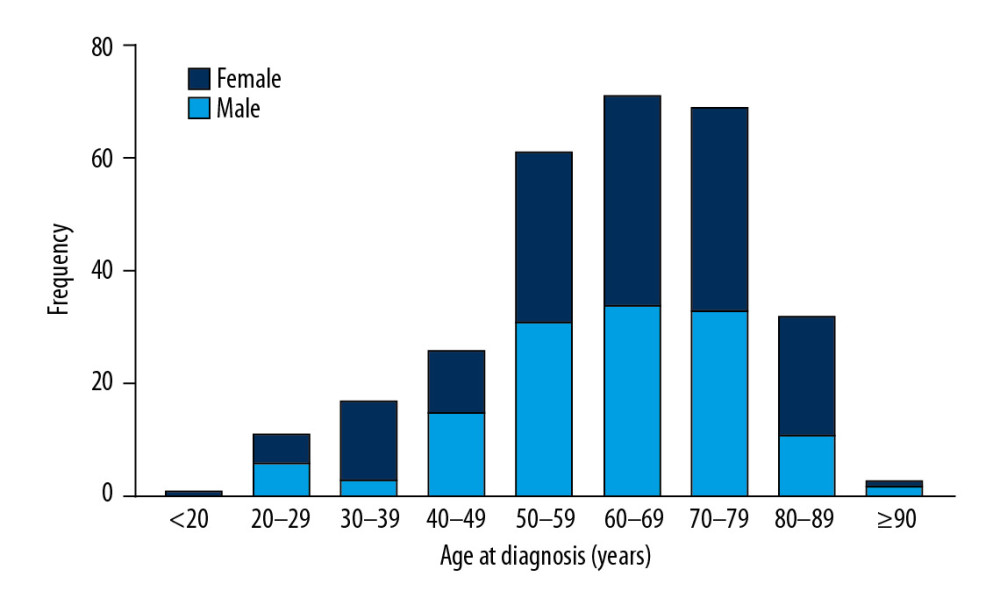 Figure 1. The distribution of age and sex of all PHNET patients.
Figure 1. The distribution of age and sex of all PHNET patients. 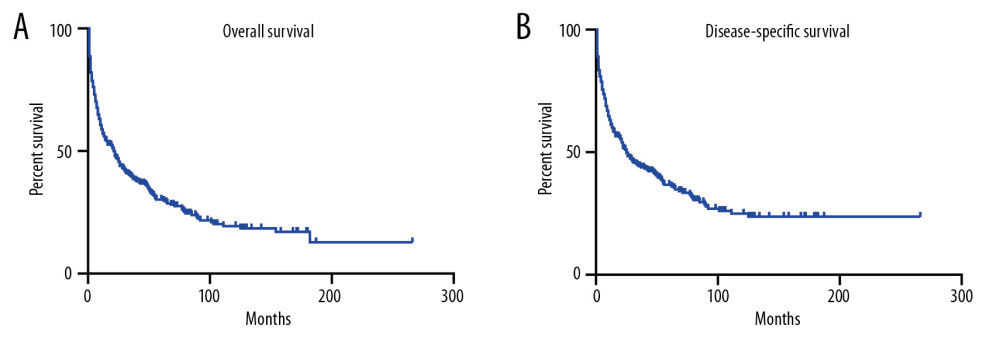 Figure 2. Kaplan-Meier analysis of overall survival (A) and disease-specific survival (B) in PHNET patients.
Figure 2. Kaplan-Meier analysis of overall survival (A) and disease-specific survival (B) in PHNET patients. 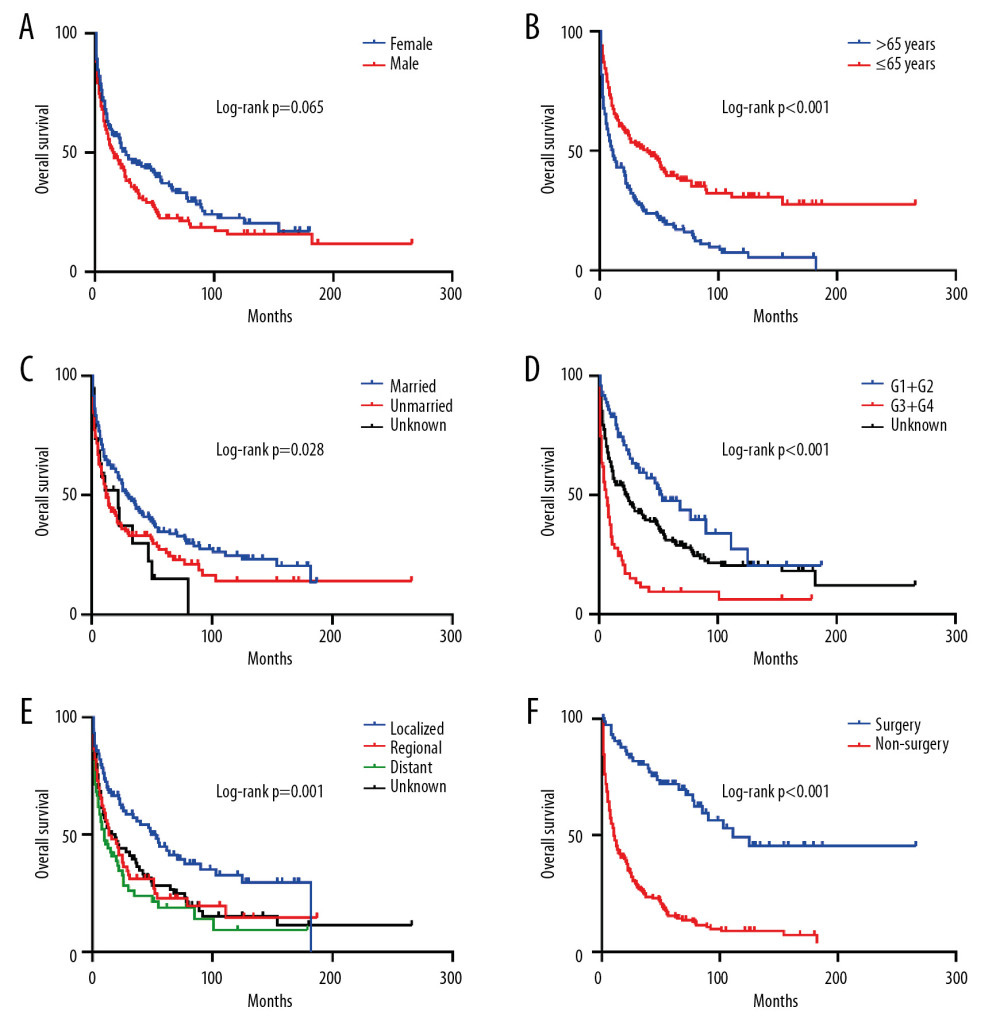 Figure 3. Kaplan-Meier analysis of overall survival in PHNET patients according to (A) Sex, (B) Age, (C) Marital status, (D) Histological grade, (E) SEER stage, (F) Tumor-directed surgery.
Figure 3. Kaplan-Meier analysis of overall survival in PHNET patients according to (A) Sex, (B) Age, (C) Marital status, (D) Histological grade, (E) SEER stage, (F) Tumor-directed surgery. 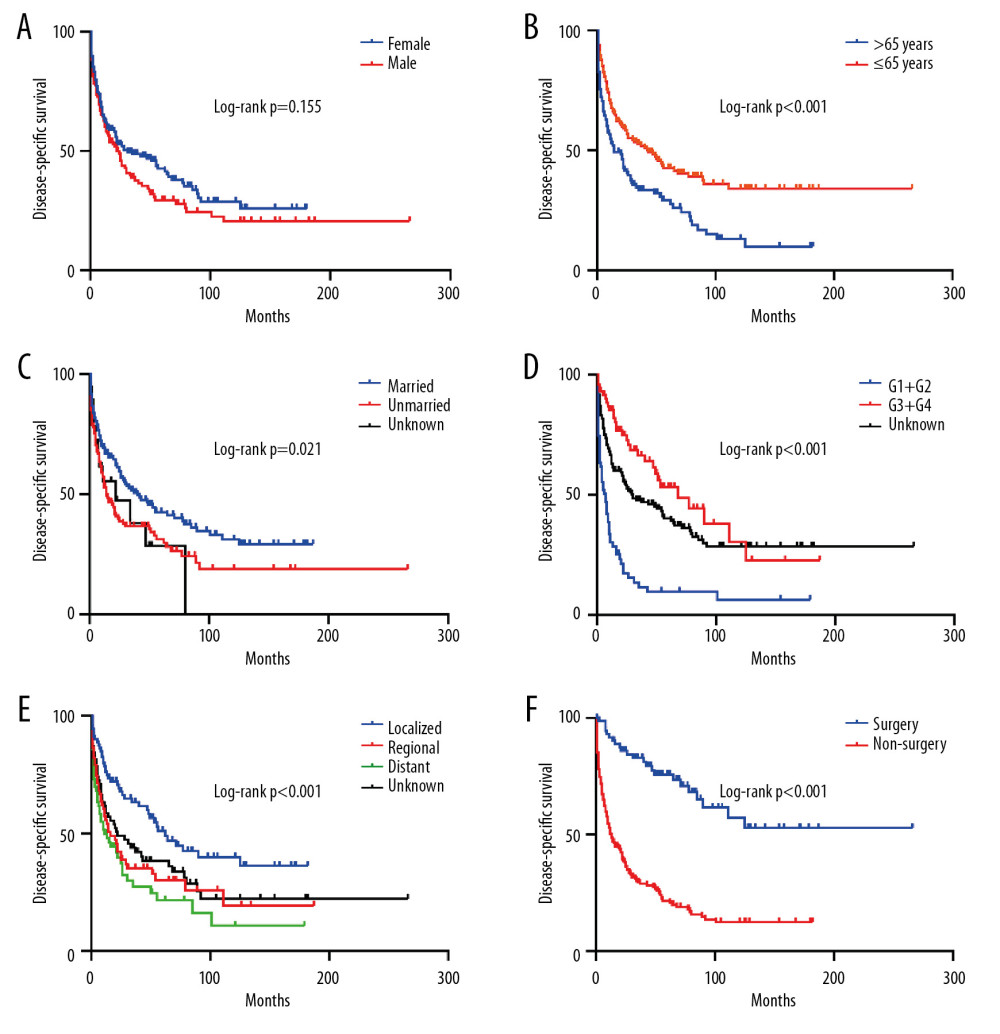 Figure 4. Kaplan-Meier analysis of disease-specific survival in PHNET patients according to (A) Sex, (B) Age, (C) Marital status, (D) Histological grade, (E) SEER stage, (F) Tumor-directed surgery.
Figure 4. Kaplan-Meier analysis of disease-specific survival in PHNET patients according to (A) Sex, (B) Age, (C) Marital status, (D) Histological grade, (E) SEER stage, (F) Tumor-directed surgery. References
1. Cives M, Strosberg JR, Gastroenteropancreatic neuroendocrine tumors: Cancer J Clin, 2018; 68; 471-87
2. Dasari A, Shen C, Halperin D, Trends in the incidence, prevalence, and survival outcomes in patients with neuroendocrine tumors in the United States: JAMA Oncol, 2017; 3; 1335-42
3. Frilling A, Åkerström G, Falconi M, Neuroendocrine tumor disease: An evolving landscape: Endocr Relat Cancer, 2012; 19; R163-85
4. Quartey , Primary hepatic neuroendocrine tumor: What do we know now?: World J Oncol, 2011; 2; 209-16
5. Edmonson HA, Tumors of the liver and intrahepatic bile ducts: Atlas Tumor Pathol, 1958; 7; 129-31
6. Park CH, Chung JW, Jang SJ, Clinical features and outcomes of primary hepatic neuroendocrine carcinomas: J Gastroenterol Hepatol, 2012; 27; 1306-11
7. Derouich H, Haddad F, Moukhlissi M, Hepatic primary neuroendocrine carcinoma: About a new case: Pan Afr Med J, 2015; 20; 254
8. Yang K, Cheng YS, Yang JJ, Primary hepatic neuroendocrine tumor with multiple liver metastases: A case report with review of the literature: World J Gastroenterol, 2015; 21; 3132-38
9. Li W, Zhuang BW, Wang Z, Case report of contrast-enhanced ultrasound features of primary hepatic neuroendocrine tumor: Med (United States), 2016; 95; e3450
10. Morishita A, Yoneyama H, Nomura T, Primary hepatic neuroendocrine tumor: A case report: Mol Clin Oncol, 2016; 4; 954-56
11. Ibrahim ME, Abadeer K, Zhai Q (Jim), Nassar A, Primary hepatic neuroendocrine tumor with unusual thyroid follicular-like morphologic characteristics: Case Rep Pathol, 2017; 2017 7931975
12. DeLuzio MR, Barbieri AL, Israel G, Emre S, Two cases of primary hepatic neuroendocrine tumors and a review of the current literature: Ann Hepatol, 2017; 16; 621-29
13. Chen RW, Qiu MJ, Chen Y, Analysis of the clinicopathological features and prognostic factors of primary hepatic neuroendocrine tumors: Oncol Lett, 2018; 15; 8604-10
14. Shi C, Zhao Q, Dai B, Primary hepatic neuroendocrine neoplasm long-time surgical outcome and prognosis: Med (United States), 2018; 97; e11764
15. Qu C, Qu LL, Zhu CZ, Treatment of primary hepatic neuroendocrine tumors with associating liver partition and portal vein ligation for staged hepatectomy (ALPPS): A case report and literature review: Med (United States), 2018; 97; e12408
16. Meng XF, Pan YW, Wang ZB, Duan WD, Primary hepatic neuroendocrine tumor case with a preoperative course of 26 years: A case report and literature review: World J Gastroenterol, 2018; 24; 2640-46
17. Zhao ZM, Wang J, Ugwuowo UC, Primary hepatic neuroendocrine carcinoma: Report of two cases and literature review: BMC Clin Pathol, 2018; 18; 3
18. Oczkowicz G, Caban A, Zemczak A, Liver transplantation as an option of treatment for a giant primary hepatic neuroendocrine tumour: Endokrynol Pol, 2019; 70; 520-21
19. Shah D, Mandot A, Cerejo C, The outcome of primary hepatic neuroendocrine tumors: A single-center experience: J Clin Exp Hepatol, 2019; 9; 710-15
20. Hu HX, Yu T, Primary hepatic neuroendocrine tumors: A case report: Med (United States), 2019; 98; e18278
21. Jung J, Hwang S, Hong SM, Long-term postresection prognosis of primary neuroendocrine tumors of the liver: Ann Surg Treat Res, 2019; 97; 176-83
22. Huang YQ, Xu F, Yang JM, Huang B, Primary hepatic neuroendocrine carcinoma: Clinical analysis of 11 cases: Hepatobiliary Pancreat Dis Int, 2010; 9; 44-48
23. Li R, Tang CL, Yang D, Primary hepatic neuroendocrine tumors: clinical characteristics and imaging features on contrast-enhanced ultrasound and computed tomography: Abdom Radiol, 2016; 41; 1767-75
24. Yang K, Cheng YS, Yang JJ, Primary hepatic neuroendocrine tumors: Multi-modal imaging features with pathological correlations: Cancer Imaging, 2017; 17; 20
25. Van Der Hoef M, Crook DW, Marincek B, Weishaupt D, Primary neuroendocrine tumors of the liver: MRI features in two cases: Abdom Imaging, 2004; 29; 77-81
26. Klimstra DS, Modlin IR, Coppola D, The pathologic classification of neuroendocrine tumors: A review of nomenclature, grading, and staging systems: Pancreas, 2010; 39; 707-12
27. Iwao M, Nakamuta M, Enjoji M, Primary hepatic carcinoid tumor: Case report and review of 53 cases: Med Sci Monit, 2001; 7; 746-50
28. Knox CD, Anderson CD, Lamps LW, Long-term survival after resection for primary hepatic carcinoid tumor: Ann Surg Oncol, 2003; 10; 1171-75
29. Sano K, Kosuge T, Yamamoto J, Primary hepatic carcinoid tumors confirmed with long-term follow-up after resection: Hepatogastroenterology, 1999; 46; 2547-50
30. Komatsuda T, Ishida H, Furukawa K, Primary carcinoid tumor of the liver: Report of a case with an emphasis on contrast-enhanced ultrasonographic findings: J Clin Ultrasound, 2005; 33; 302-4
31. Fenwick SW, Wyatt JI, Toogood GJ, Lodge JPA, Hepatic resection and transplantation for primary carcinoid tumors of the liver: Ann Surg, 2004; 239; 210-19
32. Touloumis Z, Delis SG, Triantopoulou C, Primary hepatic carcinoid; A diagnostic dilemma: A case report: Cases J, 2008; 1; 314
Figures
Figure 1. The distribution of age and sex of all PHNET patients.Figure 2. Kaplan-Meier analysis of overall survival (A) and disease-specific survival (B) in PHNET patients.Figure 3. Kaplan-Meier analysis of overall survival in PHNET patients according to (A) Sex, (B) Age, (C) Marital status, (D) Histological grade, (E) SEER stage, (F) Tumor-directed surgery.Figure 4. Kaplan-Meier analysis of disease-specific survival in PHNET patients according to (A) Sex, (B) Age, (C) Marital status, (D) Histological grade, (E) SEER stage, (F) Tumor-directed surgery. Tables
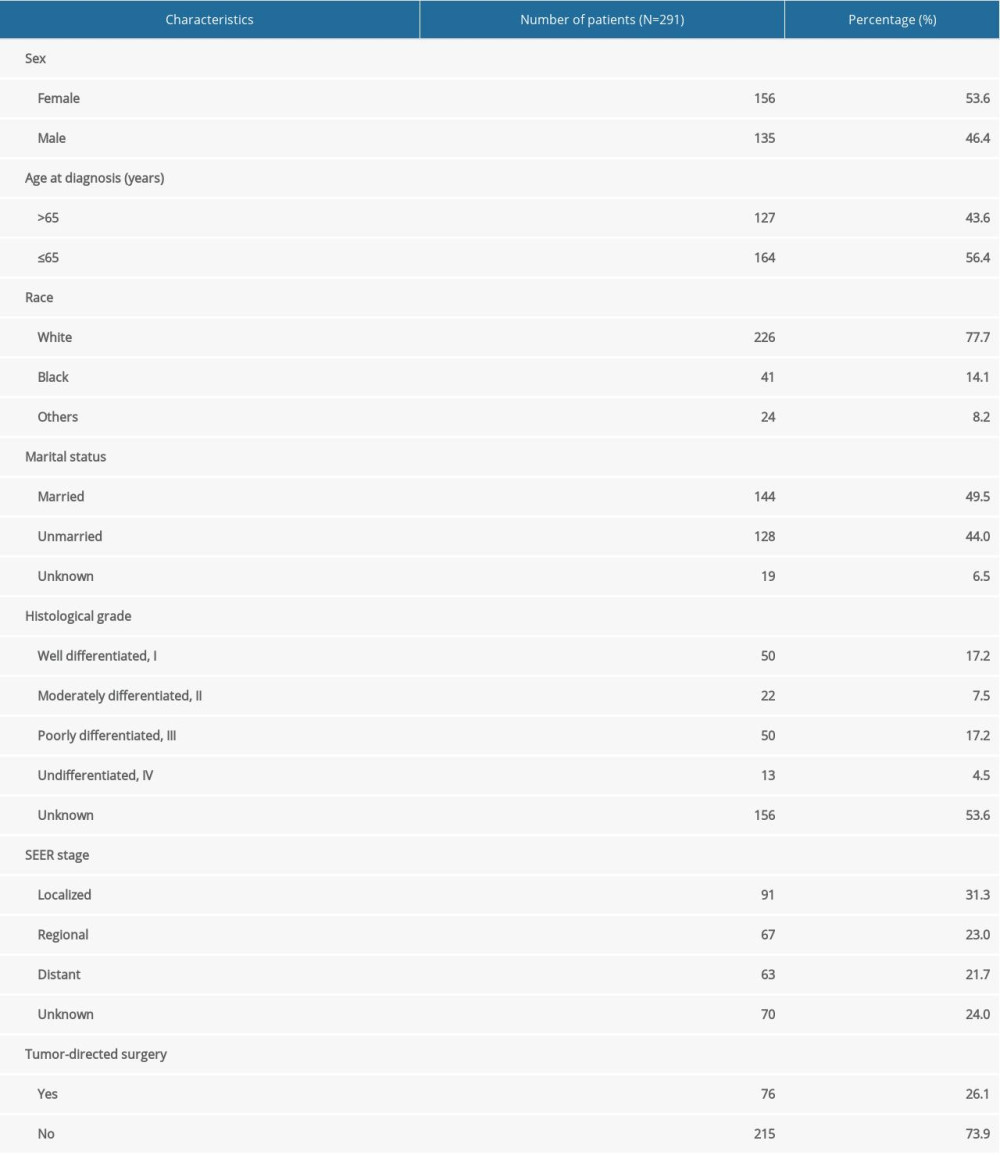 Table 1. Baseline characteristics of patients with primary hepatic neuroendocrine tumor.
Table 1. Baseline characteristics of patients with primary hepatic neuroendocrine tumor.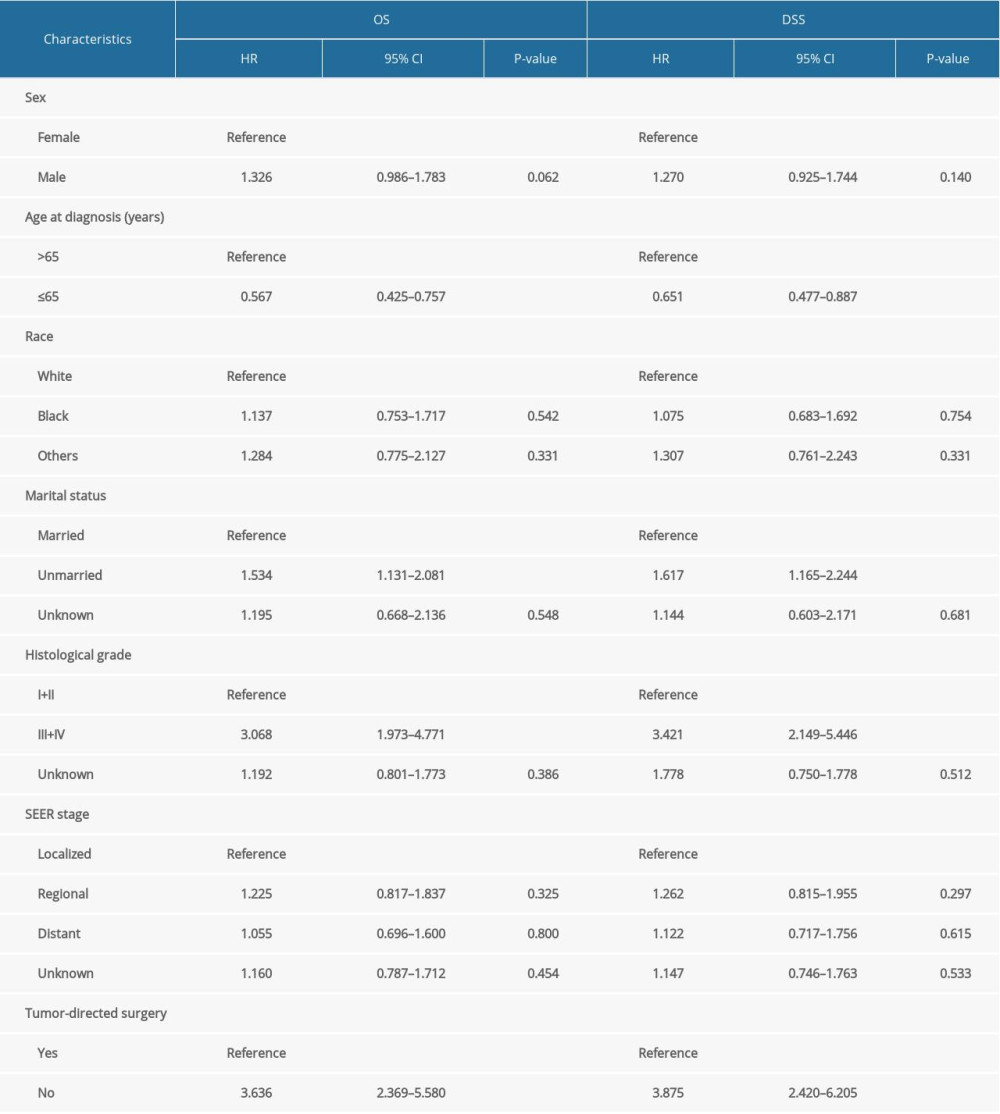 Table 2. Multivariate Cox proportional hazards regression analysis for overall survival (OS) and disease-specific survival (DSS).Table 1. Baseline characteristics of patients with primary hepatic neuroendocrine tumor.Table 2. Multivariate Cox proportional hazards regression analysis for overall survival (OS) and disease-specific survival (DSS).
Table 2. Multivariate Cox proportional hazards regression analysis for overall survival (OS) and disease-specific survival (DSS).Table 1. Baseline characteristics of patients with primary hepatic neuroendocrine tumor.Table 2. Multivariate Cox proportional hazards regression analysis for overall survival (OS) and disease-specific survival (DSS). In Press
Clinical Research
Effects of Single-Bout Endurance Exercise Intensity on Peripheral Neurotrophic Factors in Patients With Isc...Med Sci Monit In Press; DOI: 10.12659/MSM.952089
Review article
Anisodus tanguticus in Cancer Research: A Review of Traditional Use, Phytochemistry, Extraction Methods, an...Med Sci Monit In Press; DOI: 10.12659/MSM.952999
Clinical Research
Nasal Mucociliary Clearance and Its Relationship With Disease Severity in Patients With Multiple SclerosisMed Sci Monit In Press; DOI: 10.12659/MSM.952850
Clinical Research
Modified Thoracoabdominal Nerves Block Through the Perichondrial Approach vs Subcostal Transversus Abdomini...Med Sci Monit In Press; DOI: 10.12659/MSM.953976
Most Viewed Current Articles
17 Jan 2024 : Review article 14,176,570
Vaccination Guidelines for Pregnant Women: Addressing COVID-19 and the Omicron VariantDOI :10.12659/MSM.942799
Med Sci Monit 2024; 30:e942799
13 Nov 2021 : Clinical Research 3,762,188
Acceptance of COVID-19 Vaccination and Its Associated Factors Among Cancer Patients Attending the Oncology ...DOI :10.12659/MSM.932788
Med Sci Monit 2021; 27:e932788
14 Dec 2022 : Clinical Research 2,466,310
Prevalence and Variability of Allergen-Specific Immunoglobulin E in Patients with Elevated Tryptase LevelsDOI :10.12659/MSM.937990
Med Sci Monit 2022; 28:e937990
16 May 2023 : Clinical Research 708,927
Electrophysiological Testing for an Auditory Processing Disorder and Reading Performance in 54 School Stude...DOI :10.12659/MSM.940387
Med Sci Monit 2023; 29:e940387