13 December 2020: Review Articles
Does Virus-Receptor Interplay Influence Human Coronaviruses Infection Outcome?
Jolanta Bratosiewicz-Wąsik1ABEF*, Tomasz J. Wąsik2ABEFGDOI: 10.12659/MSM.928572
Med Sci Monit 2020; 26:e928572






Abstract
ABSTRACT: Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) is the third (following SARS-CoV and Middle East Respiratory Syndrome-CoV) zoonotic coronavirus that has crossed the species barrier in the 21st century, resulting in the development of serious human infection. The punishing effect of the recent outbreak of pandemic disease termed COVID-19 (coronavirus disease-19) caused by SARS-CoV-2 impelled us to gather the facts about the nature of coronaviruses. First, we introduce the basic information about coronavirus taxonomy, structure, and replication process to create the basis for more advanced consideration. In the following part of this review, we focused on interactions between the virus and the receptor on the host cell, as this stage is the critical process determining the species and tissue tropism, as well as clinical course of infection. We also illuminate the molecular basis of the strategy used by coronaviruses to cross the species barrier. We give special attention to the cellular receptor’s interaction with S protein of different CoVs (dipeptidyl peptidase IV and angiotensin-converting enzyme 2), as well as the cellular proteases involved in proteolysis of this protein. These factors determine the virus entry and replication; thus, even fine quantitative or qualitative differences in their expression may crucially affect outcomes of infection. Understanding virus biology and characterization of the host factors involved in coronavirus transmission and pathogenesis may offer novel options for development of efficient therapeutic and preventive strategies.
Keywords: Angiotensin-Converting Enzyme Inhibitors, Dipeptidyl Peptidase 4, SARS Virus, Angiotensin-Converting Enzyme 2, Coronavirus Infections, host-pathogen interactions, Middle East Respiratory Syndrome Coronavirus, Pandemics, SARS-CoV-2, Species Specificity, Spike Glycoprotein, Coronavirus, Virus Internalization, Virus Replication, Zoonoses
Background
Ever since December 2019, the world’s attention has been focused on the novel coronavirus (CoV) outbreak causing an epidemic of pneumonia in humans, spreading from Wuhan in central China throughout the world [1]. This is the third zoonotic coronavirus in the 21st century that has crossed the species barrier resulting in the development of serious human infection. The Severe Acute Respiratory Syndrome (SARS) outbreak occurred in 2002–2003 in Guangdong Province in China and was the first severe human disease caused by a coronavirus. Severe acute respiratory syndrome coronavirus (SARS-CoV) infected 8096 people and caused 774 deaths worldwide during the epidemic, resulting in a mortality rate of 9.5% [2]. In 2012, a novel human coronavirus named Middle East Respiratory Syndrome-CoV (MERS-CoV) emerged in Saudi Arabia, leading to 2494 laboratory-confirmed cases and 858 deaths, yielding a mortality rate of 34% [3]. Recently, a new coronavirus named Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) emerged in Wuhan, China as a cause of severe respiratory infection termed coronavirus disease-19 (COVID-19). As of September 13th, 28 637 952 laboratory-confirmed cases of COVID-19 have been reported, with 917 417 deaths, primarily in China as well as in 215 countries worldwide [4].
All 3 of these diseases mentioned above were triggered by coronaviruses newly emerged from a zoonotic source. The collected data on epidemiology, genetic evolution, and phylogenesis indicate that they originated in bats and most likely used domestic animals, such as cows, pigs, and camels, as intermediate hosts that enabled transmission from natural hosts to humans. Molecular and antibody surveillance identified wild animals, palm civets, and raccoon dogs, sold in the wet markets in Asia, as intermediary reservoirs of coronaviruses [5].
In addition to SARS-CoV, MERS-CoV, and SARS-CoV-2, which cause mild to severe disease in humans, 4 other human coronaviruses (HCoV-229E, HCoV-OC43, HCoV-NL63, and HCoV-HKU1) are circulating in humans and causing upper respiratory tract infections that manifest chiefly as the common cold in immunocompetent hosts, although severe infections in infants, elderly, and immunocompromised patients are observed. The molecular evolution of human coronaviruses (HCoVs) genomes and phylogenetic reconstruction provided important evidence that these viruses also originated in animals: HCoV-OC43 and HCoV-HKU1 are thought to have a rodent origin, whereas HCoV-229E and HCoV-NL63 likely originated from bats [6]. These CoVs are well adapted to humans and are widespread in the human population, and none of them have been found to be maintained within an animal reservoir [7].
Taxonomy, Morphology, and Replication
According to the International Committee for Taxonomy of Viruses, coronaviruses belong to the subfamily
CoVs are enveloped viruses possessing nonsegmented, single-stranded, positive-sense RNA genomes approximately 26–32 kb in size [9,10]. Two large overlapping open reading frames (ORFs), ORF 1a and ORF 1b, encoding 15 or 16 nonstructural proteins, occupy two-thirds of the genome at the 5′-terminus, and a one-third of the genome at the 3′-terminus encodes 4 common structural proteins in the gene order of spike (S), envelope (E), membrane (M), and nucleocapsid (N) [11,12] (Figure 1). Genomic RNA and phosphorylated nucleocapsid protein (N) form a helical nucleocapsid enveloped by a phospholipid bilayer. Two types of spike proteins – glycoprotein homotrimer (S) existing in all CoVs and the hemagglutinin-esterase (HE) in some beta-CoVs – are incorporated into the membrane. During infection, glycoprotein S is cleaved by a host cell protease into 2 separate peptides named S1 and S2. S1 is responsible for receptor binding, while S2 forms the stalk of the spike molecule [13]. Two additional proteins, which are responsible for virus assembly or virulence promotion – membrane (M) protein and envelope (E) protein – are located in the virus phospholipid bilayer [12,14–17].
S protein combines 2 biological functions: it binds to the specific surface receptor on the host cell determining the cellular tropism of the virus and mediates fusion between the viral envelope and a target cell membrane. Therefore, spike protein warrants further research. The N-terminal part of S protein is a signal peptide, which funnels protein for importation into the endoplasmic reticulum. S protein can be further divided into an N-terminal S1 subunit containing the receptor-binding domain (RBD) and an S2 subunit including the fusion peptide (FP), internal fusion peptide (IFP), and heptad repeat 1/2 (HP1/2) motifs as well as transmembrane domain (TM) [18]. The proteolytic cleavage of the CoV S protein required for the receptor recognition and for membrane fusion seems to be more complex than the proteolysis of the envelope proteins of viruses such as influenza A virus or human immunodeficiency virus [19,20]. Two cleavage sites were recognized in CoV S protein: one, for cleavage, referred to as priming, is located on the interface between S1 and S2 subunits (S1/S2 site); and the second, so called triggering, is located near the N terminus of the fusion peptide (S2’ site). Several enzymes can be engaged in proteolytic processing of spike protein. The pH-dependent cysteine protease cathepsin L, TMPRSS2 (transmembrane serine protease 2), and the serine protease furin can cut S proteins during viral entry into the target cell, and furin can cleave CoV S protein in the infected cells. As these proteases are localized at different sites of cells, they determine the cellular location of membrane fusion. Specifically, TMPRSS2 is expressed at the cell surface, so it cleaves S protein at this site (“early” entry), whereas cathepsin L present in endosomes cleaves S protein upon viral uptake into these vesicles (“late” entry). This proteolytic processing seems to be necessary to trigger the S protein, which overcomes the energy barrier associated with the membrane’s fusion [21,22]. The proteolytic cascade of MERS-CoV begins shortly after virus morphogenesis in virus-producing cells. Extensive S1/S2 cleavage at this stage allows for a similarly extensive S2’ cleavage shortly after virus binding to receptors in virus-target cells, which leads to membrane fusion triggered by early-acting proteases. However, for several CoVs, and for some MERS-CoV variants, the proteolytic cascade begins when viruses bind target cells, and then ends much later after virus endocytosis and late-acting intra-endosomal proteolysis activate a sufficient number of adjacent S proteins into fusion competence. The starting and ending points of this proteolytic cascade are correlated with cell tropism. Thus, knowledge of the proteolytic processing of membrane fusion proteins and host proteases regulating virus infections can be used to predict viral tropism and pathogenesis, and can also reveal antiviral strategies [23]. The proteolytic processing of S protein is even more fiendish, as during the triggering step, tetraspanins (CD9 for MERS-CoV) come into play. CD9 was found as an agent of DPP4 (dipeptidyl peptidase 4) and TMPR (transmembrane protease) colocalization: by linking the receptors and TMPR proteases at or near the plasma membrane, CD9 facilitated early CoV entry into target cells. The early entry route is far more likely to result in productive infection, whereas late entry appears to be a lower-efficiency, last-chance infection route before virus destruction in lysosomes [24].
The SARS-CoV-2 spike protein is so far the only known viral target of neutralizing antibodies; therefore, it has been chosen as the prime target for vaccine development. Recently, the immunoinformatics approach was used by Samad et al. to formulate a vaccine against SARS-CoV-2. By a series of computational approaches, they discovered potential T- and B-cell epitopes in S protein of SARS-CoV-2, which were embedded into a multi-epitope vaccine. Importantly, these epitopes present in the vaccine were able to strongly bind with the immune receptor TLR4 (toll-like receptor 4, known as CD284 - cluster of differentiation 284) and thus elicit robust immune response upon SARS-CoV-2 infection [25].
Replication of the CoVs is initiated by the attachment to the specific host cellular receptors via viral spike (S) protein. Interaction between S protein and cellular receptor is possible after S protein is cleaved into the S1 and S2 subunits, which remain non-covalently bound in the prefusion conformation. For all CoVs, S protein is further cleaved by host proteases at the S2’ site located immediately upstream of the fusion domain. It is believed that this cleavage activates the protein for membrane fusion via extensive irreversible conformational changes, which in turn facilitate fusion between viral and cell membrane and ultimately release the viral genome into the cytoplasm. The next step in the coronavirus lifecycle is the translation of ORF1a and ORF1b into polyproteins pp1a and pp1ab, which are sequentially cleaved to release 16 or sometimes 15 functional nonstructural proteins (Table 1) [26–29], constituting the replication-transcription complex (RTC) [12,30]. RTC is responsible for RNA replication and transcription of the sub-genomic RNAs, which serve as mRNAs for synthesis of the structural proteins S, E, and M inserted into the endoplasmic reticulum and N protein encapsidating of viral genomes. After nucleocapsid is assembled, it traffics to the endoplasmic reticulum-Golgi intermediate compartment (ERGIC) to interact with M protein and become incorporated into the viral envelope [31]. This interaction induces virion budding, transport in the vesicles, and, finally, release by exocytosis [12,32].
How can CoVs Cross the Species Barrier?
Recognition of the specific cellular receptor on the permissive cell is a critical step in the coronavirus lifecycle. The efficient virus binding to the cellular receptor via receptor-binding domain (RBD) of viral spike protein determines performance of virus replication and pathogenicity, as well as tissue tropism, and host range of the CoVs. Human coronaviruses originate from wild animal reservoirs, mostly bats and rodents, and reach humans through intermediate hosts such as Masket palm civets, raccoon dogs, and dromedary camels. The breaking of the species barrier by virus depends on achieving the ability to enter the host cell via a specific receptor with the participation of cellular proteases facilitating fusion between the viral envelope and cellular membrane. The main host cell receptors utilized by human CoVs are known: HCoV-229E binds to aminopeptidase N (APN); SARS-CoV, SARS-CoV-2, and HCoV-NL63 use angiotensin-converting enzyme 2 (ACE2); and MERS-CoV utilizes dipeptidyl peptidase 4 (DPP4) enter human cells [17,33].
Even a slight change in RBD of the S protein can influence its affinity for the cellular receptor. Yang et al. revealed that introduction of 2 single point mutations – S746R and N762A – into the genetically-related MERS-CoV bat coronavirus HKU4 spike protein fully instilled its capability to mediate viral entry into human cells. As a result of these mutations, MERS-CoV spike, but not HKU4 spike, can be activated by human endogenous proteases, which subsequently cause fusion of the viral envelope with the cell membrane. MERS-CoV S protein contains both of these mutations, which is why MERS-CoV can infect human cells [34]. Similarly, the transfer of SARS-CoV from civets to humans was enabled due to mutations N479L and T487S in the S protein’s RBD, which was sufficient to gain sustained infectivity for human cells [35]. Moreover, an altered host, tissue, or cellular tropism of the coronavirus may be achieved by genomic recombination events that result in exchange of all or a part of the gene encoding S protein. Type II feline coronavirus (FCoV) emerged inside the cat body via double recombination between type I FCoV and type II canine coronavirus (CCoV), in which the feline virus acquired a type II CCoV spike gene. As a result of the acquisition of this new S protein, the rather harmless enteric type I FCoV turned into a systemically replicating and deadly peritonitis virus [36].
The cellular tropism and pathogenesis of coronaviruses is also affected by the presence of the specific cleavage sequences within S protein targeted by cells’ proteases. It has been demonstrated that the insertion of a furin-like cleavage site in the infectious bronchitis virus (IBV) S protein resulted in higher pathogenicity [37]. Comparison of the S protein cleavage sites sequence has revealed that SARS-CoV-2 have a furin-like cleavage site absent in other CoVs of the same clade. This furin-like cleavage site is thought to provide a gain-of-function to SARS-CoV-2 for efficient spreading in the human population compared to other lineage b betacoronaviruses [38].
After achieving the ability of replication within a new species, CoVs may still acquire mutations allowing escape from neutralizing antibody or improving replication efficiency. The D614G amino acid change of SARS-CoV-2 spike protein appeared in early March 2020 and G614 has become the dominant form in the pandemic, implying that this change enhanced viral transmission. G614 variant was associated with lower RT-PCR cycle thresholds, which reflects higher upper respiratory tract viral loads. It was proposed that the D614G change would eliminate the side-chain hydrogen bond between protomers, thus possibly increasing their flexibility and altering between-protomer interactions. In addition, this substitution can modulate glycosylation at the nearby N616 site, influencing the dynamics of the spatially proximal fusion of the neighboring protomer [39,40].
Cellular Receptors for CoVs
DIPEPTIDYL PEPTIDASE IV (DPP4):
Dipeptidyl peptidase IV (DPP4), also known as CD26, was identified as a receptor for MERS-CoV by Raj et al. in 2013 by specifical co-purification of DPP4 with the receptor-binding S1 domain of the MERS-CoV spike protein from lysates of susceptible Huh-7 cells. The authors also revealed that antibodies directed against DPP4 inhibited MERS-CoV infection of primary human bronchial epithelial cells and Huh-7 cells [42]. CD26/DPP4 is a type II integral membrane protein, organized as a homodimer, with each monomer comprising a membrane-proximal α/β hydrolase domain and a membrane-distal β-propeller domain. The α/β hydrolase domain cleaves-off either X-L-proline or X-L-alanine dipeptides from various proteins such as hormones, cytokines, chemokines, neuropeptides, and vasoactive peptides. The receptor-binding domains (RBDs) of the MERS-CoV S proteins bind to human blades 4 and 5 of the 8-blade β-propeller conformation [43,44]. Eleven critical residues in DPP4, which directly contact the MERS-CoV RBD, were identified by Wang et al. and Lu et al. [43,45]. These residues are highly conserved in camelids, primates, and rabbits, which are all shown to be susceptible to MERS-CoV. In contrast, ferrets, rats, and mice resist MERS-CoV infection due to differences in some critical DPP4 residues. These data illustrate that the DPP4 structure itself can determine the host range of MERS-CoV [46].
Several lines of evidence have revealed that tissue-specific localization, levels of expression, as well as structure of DPP4 protein affect MERS-CoV transmission and pathogenesis. The localization of DPP4 expression contributes to more efficient virus spreading in dromedary camels than in humans. Differences in viral shedding in dromedary camels and humans are attributable to the ability of the virus to replicate in the upper respiratory tract. Relatively high levels of infectious virus were detected in nasal swabs of dromedaries infected with MERS-CoV, while in MERS patients the virus mainly replicates in the lower respiratory tract. In humans, DPP4 is absent in the nasal epithelium but present in the lower respiratory tract epithelium, mainly in type II pneumocytes. In contrast, DPP4 is expressed in the nasal epithelium of dromedary camels. This difference in DPP4 localization between humans and dromedary camels explains MERS-CoV tropism in these 2 species and highlights DPP4 as an essential determinant of MERS-CoV tropism [47].
The next enigma that was solved by studying the role of DPP4 in MERS-CoV pathogenesis was intraspecies variation in the clinical course of MERS-CoV infection. Clinical symptoms of MERS range from asymptomatic infection to severe pneumonia with acute respiratory distress syndrome, resulting in death [48]. This implies that some host factors can dictate the outcome of MERS-CoV infection, thus producing intraspecies variation. Two of these risk factors influencing the clinical course of MERS-CoV infection are smoking and chronic obstructive pulmonary disease (COPD). Smokers and COPD patients have been reported to be more susceptible to MERS-CoV infection. Seys et al. revealed that in smokers and subjects with COPD, both DPP4 mRNA and protein expression were significantly higher compared to never-smokers, suggesting DPP4 as a possible cause of intraspecies variation observed among MERS-CoV patients [49]. About 98% of the internal surface area of the lung is lined with alveolar type I cells, while the remainder is lined with type II cells. In healthy human lungs, DPP4 is almost exclusively expressed on type II pneumocytes, whereas in the lungs of smokers and COPD patients, DPP4 is prominently expressed on both type I and II pneumocytes, indicating upregulated expression on type I pneumocytes. DPP4 expression on type I pneumocytes can lead to damage of these cells in the lung alveoli during viral infection and ultimately to diffuse alveolar damage [46].
As mentioned earlier, the structure of the interface between DPP4 and MERS-CoV S protein was resolved on the molecular level and 15 residues in DPP4 were found to make direct contact with residues in the viral S protein [45]. Kleine-Weber et al. asked whether naturally-occurring amino acid polymorphisms in DPP4 residues making contact with MERS-CoV S have an impact on MERS-CoV entry. Out of 14 polymorphisms listed in public databases, 4 mutations (K267E, K267N, A291P, and Δ346–348) reduced S protein-driven host cell entry and replication of MERS-CoV by lowering the binding efficiency of MERS-CoV S to DPP4, suggesting that the DPP4 phenotype can affect the course of MERS-CoV infection. The DPP4 Δ346–348 variant is inefficiently transported to the cell surface, whereas K267E and A291P DPP4 polymorphisms reduced S protein binding to DPP4. Thus, the naturally-occurring polymorphisms in DPP4 can negatively impact cellular entry of MERS-CoV and thus can modulate MERS development in infected patients [50].
ANGIOTENSIN-CONVERTING ENZYME 2 (ACE2):
Angiotensin-converting enzyme 2 (ACE2) is an 805 amino acid type I transmembrane glycoprotein with an extracellular catalytic domain, with a molecular weight of approximately 120 kDa. ACE2 was found to share approximately 40% identity with the N- and C-terminal domains of ACE. ACE2 is localized predominantly in the heart, kidneys, testes, liver, and intestines, and occurs at a lower level in a wide variety of tissues, particularly the colon and lung. ACE2 belong to the M2 family of metalloproteases, and has their active site domains exposed to the extracellular surface, facilitating the metabolism of circulating peptides [51]. ACE cleaves angiotensin I (AngI), which is a decapeptide, into octapeptide angiotensin II (AngII), while ACE2 removes a single residue from AngI to yield Ang1–9 and cleaves a single residue from AngII to generate Ang1–7. In several studies, ACE2 has emerged as a potent negative regulator of the renin-angiotensin system (RAS), playing an opposing role to ACE in diverse organ systems including the heart, kidneys, and lungs. In the respiratory system, ACE, AngII, and AngII receptor type 1 (AT1R) function as lung-injury-promoting factors, whereas ACE2 protects from lung injury [52]. One of the reasons for the severe course of SARS-CoV infection with such high mortality may be marked by downregulation of ACE2 expression in the lungs, as revealed by Kuba et al. in 2005, who proved that SARS-CoV infection as well as a recombinant spike protein of SARS-CoV reduce ACE2 expression. Thus, dual action can be attributed to ACE2 in the pathogenesis of CoVs lung infection; it plays a crucial role in cellular entry and its downregulation deprives lungs of protection from injury [53] (Figure 2). Moreover, differences in downregulation of ACE2 by spike proteins of SARS and NL63 coronaviruses may elucidate the distinct clinical course of infections with these viruses, as recombinant SARS S protein binds to ACE2 and induces ACE2 shedding with higher efficiency than NL63 S protein [54]. The biophysical and structural evidence suggest that SARS-CoV-2 can bind ACE2 with a much higher affinity than SARS-CoV. This difference in the affinity of binding to ACE2 may contribute to the apparent ease with which SARS-CoV-2 can spread from human to human [55]. This is in accordance with the report by Chu et al., who revealed that SARS-CoV-2 infected and replicated in human lung tissues more efficiently than SARS-CoV [56].
Disturbance of RAS balance via binding of SARS-CoV S protein with ACE2 was shown to play a key role in the pathogenesis of lung injury through direct and indirect pathways. AngII activation induced increased pulmonary vascular permeability leading to pulmonary edema and impairing lung function [57]. Activation of AngII pathways leads to stimulation of macrophages and other immune cells [58] and enhances concentrations of inflammatory cytokines such as interleukin 6 (IL-6) and tumor necrosis factor-alpha (TNF-α) [59,60] as well as the phagocytosis of the apoptotic virus-infected epithelial cells [61]. The enhanced levels of proinflammatory mediators, the “cytokine storm”, followed by the attacking immune system results in lung failure, multiple-organ dysfunction, and death in severe cases [62].
Studies on many other viruses demonstrated that the presence of allelic variants of host receptor determined the efficiency of virus binding and thus influenced the degree of host resistance against viral infection. Hence, efforts were made to determine if genetic variants of ACE2 can moderate the outcomes of SARS-CoV infection. There are very few single-nucleotide polymorphisms (SNPs) within the coding regions of the human ACE2 gene, and most of them are observed with lower that 5% minor allele frequency. Case-control studies failed to obtain any evidence that ACE2 coding and non-coding polymorphisms were involved in SARS susceptibility or outcome in Chinese and Vietnamese populations [63,64].
Hussain et al. constructed molecular models of coding variants of ACE2 corresponding to the reported binding sites for its attachment with coronavirus spike protein to observe structural changes in the ACE2 variants and their intermolecular interactions with SARS-CoV-2 spike protein. ACE2 alleles, rs73635825 (S19P), and rs143936283 (E329G) showed noticeable variations in their intermolecular interactions with the viral spike protein. These data provide a structural basis for understanding the potential resistance against SARS-CoV-2 infection driven by ACE2 allelic variants [65]. However, it is difficult to estimate the clinical significance of these polymorphisms because the observed frequencies are relatively low: 3.3×10−3 for rs73635825 among Africans and 6.51×10−5 for rs143936283 among Europeans [66].
Recently, very detailed analysis of ACE2 expression was performed using a single-cell RNA sequencing technique (RNA-Seq). An earlier immunostaining investigation showed that in normal human lungs, ACE2 is mainly expressed by type II (AT2) and type I (AT1) alveolar epithelial cells. Zhao et al. revealed that the majority of the ACE2-expressing cells (an average of 83%) are AT2 cells. Other ACE2-expressing cells include AT1 cells, airway epithelial cells, fibroblasts, endothelial cells, and macrophages. It is especially intriguing that ACE2-positive AT2 cells also express many other proteins promoting viral reproduction and transmission. Therefore, it seems that SARS-CoV-2 has cleverly evolved to hijack this population of AT2 cells for its reproduction and transmission. Of note, male subjects have a higher ACE2-expressing cell ratio than female subjects, and the distribution of ACE2 is also more widespread in males than females. This result is highly consistent with the epidemiologic investigation showing that most of the SARS-CoV-2-infected patients were males [67]. However, analysis of 4 large datasets of normal lung tissue revealed no significant disparities in ACE2 gene expression between racial groups (Asian vs. White), age groups (>60 years vs. <60 years), or between males and females. Different conclusions may be drawn based on studies of Xie et al., who found that lung ACE2 levels decline with age in rat models, and this decline is greater in males [68]. This naturally-occurring decrease in ACE2 level in combination with downregulation of ACE2 expression caused by SARS-CoV-2 S protein may explain the severe outcome of infection in older patients. However, significantly higher ACE2 gene expression was observed in smokers’ samples compared to nonsmokers’ samples, suggesting that the smokers are more susceptible to SARS-CoV-2 infection [69,70]. These results go hand in hand with the observation that cigarette smoking causes a dose-dependent upregulation of ACE2 in rodent and human lungs. Using single-cell sequencing data, the expression of ACE2 was demonstrated in a subset of epithelial cells that line the respiratory tract, including goblet cells, club cells, and alveolar type 2 cells. Chronic smoke exposure triggers a protective expansion of mucus-secreting goblet cells and a concomitant increase in ACE2 expression. Thus, smoking history should be considered in identifying susceptible populations and standardizing treatment regimens [71].
Recently, one more aspect of organ-specific distribution of ACE2 expression was addressed by Fu et al., who detected high levels of expression in testes and in the cardiovascular and gastrointestinal systems. They suggested that SARS-CoV-2 not only attacks the lungs, but also affects other organs, particularly the testes, and in consequence severely damages male sexual development in younger males, leading to adult infertility. Moreover, the expression level of ACE2 was considered as a prognostic marker in some cancers. It has been found that high expression of ACE2 was correlated with longer survival in renal and liver cancers [72], but in lung cancers, the upregulation of ACE2 predicts poor survival of COVID-19 patients with lung malignancies [73,74].
Recently, researchers have found that ACE2 and other members of the renin-angiotensin system participate in different biological processes in various tumors. ACE2, as well as Ang1–7, were reported to inhibit the growth of lung cancer [75,76]. Moreover, ACE2 can inhibit breast cancer angiogenesis and metastasis through suppressing the isoform of a vascular endothelial growth factor/VEGF receptor 2/extracellular signal-regulated kinase (VEGFa/VEGFR2/ERK) pathway [77].
Spike Protein Cleavage
In addition to host receptor recognition, proteolytic processing of the S protein, which ultimately results in virus-cell fusion, is the second crucial step for virus tropism at the cellular, tissue, and host species level. As mentioned in the Taxonomy, Morphology, and Replication section above, S protein comprises 2 functional subunits: the S1 subunit, which is responsible for binding to the host cell receptor, and the S2 subunit, which ensures fusion of the viral and cellular membranes. Prior to and/or during endocytic uptake, the CoV S protein is proteolytically processed. The spike protein may contain 2 proteolytic cleavage sites. One of the cleavage sites is located at the boundary between the S1 and S2 subunits (S1/S2 cleavage site), while the other cleavage site is located immediately upstream of the first of 2 fusion peptides (S2 cleavage site) located within the S2 domain. The specific proteolytic cleavage requirements of the S protein at the S1/S2 boundary and particularly at the S2’ site may furthermore determine the intracellular site of fusion. It has become evident that the protease expression profile of host cells may form an additional determinant of the host cell tropism of coronaviruses [78]. TMPRSS2 has been found to perform both SARS-CoV and SARS-CoV-2 S protein priming and hemagglutinin cleavage to activate internalization of the influenza virus A. It has been shown that TMPRSS2 inhibitor blocks entry and might be a treatment option [41].
Immunohistochemistry studies revealed that the TMPRSS2 protein is expressed on type II alveolar cells and alveolar macrophages, and that there is no expression in type I alveolar cells that form the respiratory surface [79,80]. Importantly, patients who carried a single-nucleotide polymorphism associated with higher TMPRSS2 expression were more susceptible to influenza virus infection [81].
Interestingly, heightened androgen receptor signaling induces the fusion of
Conclusions
The emergence of the third coronavirus outbreak during the 21st century made us aware of how easily these viruses can mutate to cross the species barriers and cause outbreaks in both humans and animals. It is necessary to understand the detailed mechanisms of the virus life cycle and interactions with the host proteins to develop efficient therapeutic and preventive strategies for these viruses that until very recently were considered harmless. On the other hand, as the host factors determine the virus entry and replication, even slight quantitative or qualitative differences in their expression may be crucial for the outcome of infection. Thus, recognition of the host receptors involved in the virus life cycle is equally important for epidemic overcome.
Figures
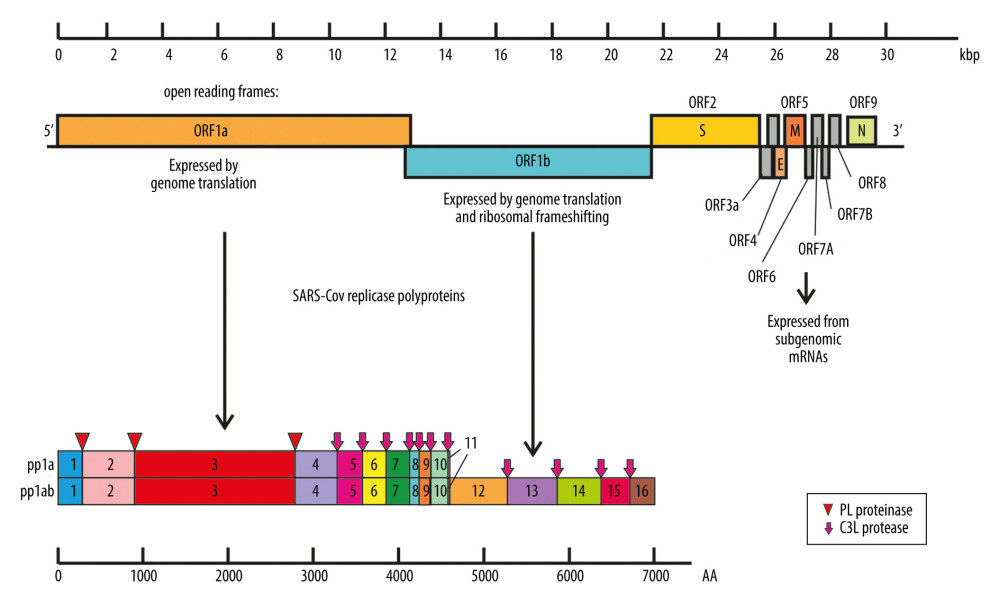 Figure 1. Coronaviruses genome organization. Localized from the 5′ end, there are 2 open reading frames (ORF) – ORF1a and ORF1b – which encode polyproteins pp1a and pp1ab that are sequentially cleaved to release 16 or sometimes 15 functional nonstructural proteins constituting the replication-transcription complex (RTC). ORFs localized by the 3′ end encode structural and regulatory proteins: spike (S), ORF3A, envelope (E), membrane (M), ORF5, ORF6, ORF7A, ORF7B, ORF8, and nucleocapsid (N). The bottom panel shows the proteolytic processing of the pp1a and pp1ab polyproteins. The numbers of polyproteins pp1a and pp1ab segments correspond to nonstructural proteins: 3 – PLpro, papain-like proteinase; 5–3CLpro, 3C-like protease; 12 – RdRp, RNA-dependent RNA polymerase; 13 – HEL1, superfamily 1 helicase; 14 – ExoN, exoribonuclease, N7-MT, N7-methyl transferase; 15 – endoU, uridylate-specific endoribonuclease; and 16–2′-O-MT, 2′-O-methyl transferase. Functions of remaining nonstructural proteins are described in Table 1.
Figure 1. Coronaviruses genome organization. Localized from the 5′ end, there are 2 open reading frames (ORF) – ORF1a and ORF1b – which encode polyproteins pp1a and pp1ab that are sequentially cleaved to release 16 or sometimes 15 functional nonstructural proteins constituting the replication-transcription complex (RTC). ORFs localized by the 3′ end encode structural and regulatory proteins: spike (S), ORF3A, envelope (E), membrane (M), ORF5, ORF6, ORF7A, ORF7B, ORF8, and nucleocapsid (N). The bottom panel shows the proteolytic processing of the pp1a and pp1ab polyproteins. The numbers of polyproteins pp1a and pp1ab segments correspond to nonstructural proteins: 3 – PLpro, papain-like proteinase; 5–3CLpro, 3C-like protease; 12 – RdRp, RNA-dependent RNA polymerase; 13 – HEL1, superfamily 1 helicase; 14 – ExoN, exoribonuclease, N7-MT, N7-methyl transferase; 15 – endoU, uridylate-specific endoribonuclease; and 16–2′-O-MT, 2′-O-methyl transferase. Functions of remaining nonstructural proteins are described in Table 1. 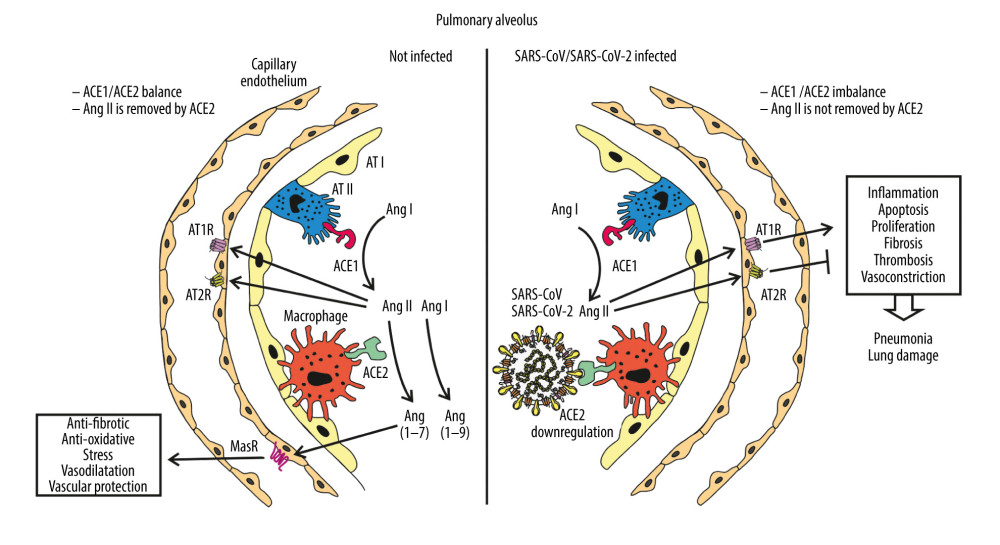 Figure 2. Schematic diagram of alveolus during normal homeostasis (left panel) characterized by a balanced ACE1/ACE2 pathway, and under SARS-CoV-2 infection condition (right panel) where ACE2 receptor is downregulated by SARS-CoV-2 and increasing ACE1/ACE2 unbalance causes the renin-angiotensin system (RAS) hyper-activation leading to lung injury. AT I, AT II – alveolar type I, II epithelial cell (type I, II pneumocyte); ACE1, ACE2 – angiotensin-converting enzyme 1, 2; MasR – Mas receptor; Ang I, II – angiotensin I, II; AT1R, AT2R – angiotensin II type 1, 2 receptor.
Figure 2. Schematic diagram of alveolus during normal homeostasis (left panel) characterized by a balanced ACE1/ACE2 pathway, and under SARS-CoV-2 infection condition (right panel) where ACE2 receptor is downregulated by SARS-CoV-2 and increasing ACE1/ACE2 unbalance causes the renin-angiotensin system (RAS) hyper-activation leading to lung injury. AT I, AT II – alveolar type I, II epithelial cell (type I, II pneumocyte); ACE1, ACE2 – angiotensin-converting enzyme 1, 2; MasR – Mas receptor; Ang I, II – angiotensin I, II; AT1R, AT2R – angiotensin II type 1, 2 receptor. 
References
1. Huang C, Wang Y, Li X, Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China: Lancet, 2020; 395(10223); 497-506
2. World Health Organization: Summary of probable SARS cases with onset of illness from 1 November 2002 to 31 July 2003 https://www.who.int/csr/sars/country/table2004_04_21/en/
3. World Health Organization: Middle East respiratory syndrome coronavirus (MERS-CoV) November, 2019 http://www.who.int/emergencies/mers-cov/en/
4. World Health Organization: Coronavirus disease (COVID-19) weekly epidemiological update https://www.who.int/docs/default-source/coronaviruse/situation-reports/20200831-weekly-epi-update-3.pdf?sfvrsn=d7032a2a_4
5. Gralinski LE, Menachery VD, Return of the coronavirus: 2019-nCoV: Viruses, 2020; 12; 135
6. Forni D, Cagliani R, Clerici M, Molecular evolution of human coronavirus genomes: Trends Microbiol, 2017; 25; 35-48
7. Su S, Wong G, Shi W, Epidemiology, genetic recombination, and pathogenesis of coronaviruses: Trends Microbiol, 2016; 24; 490-502
8. https://talk.ictvonline.org/taxonomy/
9. Gorbalenya AE, Enjuanes L, Ziebuhr J, Snijder EJ, Nidovirales: Eolving the largest RNA virus genome: Virus Res, 2006; 117(1); 17-37
10. Chen Y, Liu Q, Guo D, Emerging coronaviruses: Genome structure, replication, and pathogenesis: J Med Virol, 2020; 92; 418-23
11. Sawicki SG, Sawicki DL, Siddell SG, A contemporary view of coronavirus transcription: J Virol, 2007; 81(1); 20-29
12. Fehr AR, Perlman S, Coronaviruses: An overview of their replication and pathogenesis: Methods Mol Biol, 2015; 1282; 1-23
13. Belouzard S, Chu VC, Whittaker GR, Activation of the SARS coronavirus spike protein via sequential proteolytic cleavage at two distinct sites: Proc Natl Acad Sci USA, 2009; 106(14); 5871-76
14. Beniac DR, Andonov A, Grudeski E, Booth TF, Architecture of the SARS coronavirus prefusion spike: Nat Struct Mol Biol, 2006; 13(8); 751-52
15. Neuman BW, Kiss G, Kunding AH, A structural analysis of M protein in coronavirus assembly and morphology: J Struct Biol, 2011; 174(1); 11-22
16. Nal B, Differential maturation and subcellular localization of severe acute respiratory syndrome coronavirus surface proteins S, M and E: J Gen Virol, 2005; 86(Pt 5); 1423-34
17. Li G, Fan Y, Lai Y, Coronavirus infections and immune responses: J Med Virol, 2020; 92; 424-32
18. Heald-Sargent T, Gallagher T, Ready, set, fuse! The coronavirus spike protein and acquisition of fusion competence: Viruses, 2012; 4(4); 557-80
19. Hallenberger S, Bosch V, Angliker H, Inhibition of furin-mediated cleavage activation of HIV-1 glycoprotein gp160: Nature, 1992; 360(6402); 358-61
20. Stieneke-Gröber A, Vey M, Angliker H, Influenza virus hemagglutinin with multibasic cleavage site is activated by furin, a subtilisin-like endoprotease: EMBO J, 1992; 11(7); 2407-14
21. Burkard C, Verheije MH, Wicht O, Coronavirus cell entry occurs through the endo-/lysosomal pathway in a proteolysis-dependent manner: PLoS Pathog, 2014; 10(11); e1004502
22. Hoffmann M, Hofmann-Winkler H, Pöhlmann S, Priming time: How cellular proteases arm coronavirus spike proteins: Activation of viruses by host proteases, 2018; 71-98, Springer International Publishing AG, part of Springer Nature
23. Park JE, Li K, Barlan A, Proteolytic processing of Middle East respiratory syndrome coronavirus spikes expands virus tropism: Proc Natl Acad Sci USA, 2016; 113; 12262-67
24. Hantak MP, Qing E, Earnest JT, Gallagher T, Tetraspanins: Architects of viral entry and exit platforms: J Virol, 2019; 93; e01429-17
25. Samad A, Ahammad F, Nain Z, Designing a multi-epitope vaccine against SARS-CoV-2: An immunoinformatics approach: J Biomol Struct Dyn, 2020 [Online ahead of print]
26. Yoshimoto FK, The proteins of severe acute respiratory syndrome coronavirus-2 (SARS CoV-2 or n-COV19), the cause of COVID-19: Protein J, 2020; 39(3); 198-216
27. Neuman BW, Buchmeier MJ, Supramolecular architecture of the coronavirus particle: Adv Virus Res, 2016; 96; 1-27
28. Snijder EJ, Decroly E, Ziebuhr J, The nonstructural proteins directing coronavirus RNA synthesis and processing: Adv Virus Res, 2016; 96; 59-126
29. Narayanan K, Ramirez SI, Lokugamage KG, Makino S, Coronavirus nonstructural protein 1: Common and distinct functions in the regulation of host and viral gene expression: Virus Res, 2015; 202; 89-100
30. Walls AC, Park YJ, Tortorici MA, Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein: Cell, 2020; 181(2); 281-92
31. de Haan CA, Rottier PJ, Molecular interactions in the assembly of coronaviruses: Adv Virus Res, 2005; 64; 165-230
32. Lim YX, Ng YL, Tam JP, Liu DX, Human coronaviruses: A review of virus-host interactions: Diseases, 2016; 4(3); 26
33. de Wilde AH, Snijder EJ, Kikkert M, van Hemert MJ, Host factors in coronavirus replication: Curr Top Microbiol Immunol, 2018; 419; 1-42
34. Yang Y, Liu C, Du L, Two mutations were critical for bat-to-human transmission of Middle East respiratory syndrome coronavirus: J Virol, 2015; 89; 9119-23
35. Li F, Structural analysis of major species barriers between humans and palm civets for severe acute respiratory syndrome coronavirus infections: J Virol, 2008; 82; 6984-91
36. Terada Y, Matsui N, Noguchi K, Emergence of pathogenic coronaviruses in cats by homologous recombination between feline and canine coronaviruses: PLoS One, 2014; 9(9); e106534
37. Cheng J, Zhao Y, Xu G, The S2 subunit of QX-type infectious bronchitis coronavirus spike protein is an essential determinant of neurotropism: Viruses, 2019; 11(10); 972
38. Coutard B, Valle C, de Lamballerie X, The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade: Antiviral Res, 2020; 176; 104742
39. Korber B, Fischer WM, Gnanakaran S, Tracking changes in SARS-CoV-2 spike: Evidence that D614G increases infectivity of the COVID-19 virus: Cell, 2020; 182(4); 812-27
40. Zhang L, Jackson CB, Mou H, The D614G mutation in the SARS-CoV-2 spike protein reduces S1 shedding and increases infectivity: bioRxiv, 2020; 2020 148726
41. Hoffmann M, Kleine-Weber H, Schroeder S, SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor: Cell, 2020; 181(2); 271-80
42. Raj VS, Mou H, Smits SL, Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus-EMC: Nature, 2013; 495; 251-54
43. Lu G, Hu Y, Wang Q, Molecular basis of binding between novel human coronavirus MERS-CoV and its receptor CD26: Nature, 2013; 500; 227-31
44. Boonacker E, Van Noorden CJ, The multifunctional or moonlighting protein CD26/DPPIV: Eur J Cell Biol, 2003; 82; 53-73
45. Wang N, Shi X, Jiang L, Structure of MERS-CoV spike receptor-binding domain complexed with human receptor DPP4: Cell Res, 2013; 23; 986-93
46. Widagdo W, Sooksawasdi Na Ayudhya S, Hundie GB, Haagmans BL, Host determinants of MERS-CoV transmission and pathogenesis: Viruses, 2019; 11(3); 280
47. Widagdo W, Raj VS, Schipper D, Differential expression of the Middle East respiratory syndrome coronavirus receptor in the upper respiratory tracts of humans and dromedary camels: J Virol, 2016; 90; 4838-42
48. Zumla A, Hui DS, Perlman S, Middle East respiratory syndrome: Lancet, 2015; 3; 60454-58
49. Seys LJM, Widagdo W, Verhamme FM, DPP4, the Middle East respiratory syndrome coronavirus receptor, is upregulated in lungs of smokers and chronic obstructive pulmonary disease patients: Clin Infect Dis, 2018; 66; 45-53
50. Kleine-Weber H, Schroeder S, Krüger N, Polymorphisms in dipeptidyl peptidase 4 reduce host cell entry of Middle East respiratory syndrome coronavirus: Emerg Microbes Infect, 2020; 9; 155-68
51. Kuba K, Imai Y, Ohto-Nakanishi T, Penninger JM, Trilogy of ACE2: A peptidase in the renin-angiotensin system, a SARS receptor, and a partner for amino acid transporters: Pharmacol Ther, 2010; 128; 119-28
52. Imai Y, Kuba K, Ohto-Nakanishi T, Penninger JM, Angiotensin-converting enzyme 2 (ACE2) in disease pathogenesis: Circ J, 2010; 74; 405-10
53. Kuba K, Imai Y, Rao S, A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury: Nat Med, 2005; 11; 875-79
54. Glowacka I, Bertram S, Herzog P, Differential downregulation of ACE2 by the spike proteins of severe acute respiratory syndrome coronavirus and human coronavirus NL63: J Virol, 2010; 84; 1198-205
55. Wrapp D, Wang N, Corbett , Cryo-EM Structure of the 2019-nCoV spike in the prefusion conformation: Science, 2020; 367(6483); 1260-63
56. Chu H, Chan JF, Wang Y: Clin Infect Dis, 2020; 71(6); 1400-9
57. Imai Y, Kuba K, Rao S, Angiotensin-converting enzyme 2 protects from severe acute lung failure: Nature, 2005; 436(7047); 112-16
58. Bernstein KE, Khan Z, Giani JF, Angiotensin-converting enzyme in innate and adaptive immunity: Nat Rev Nephrol, 2018; 14; 325-36
59. Lee YB, Nagai A, Kim SU, Cytokines, chemokines, and cytokine receptors in human microglia: J Neurosci Res, 2002; 69; 94-103
60. Yamamoto S, Yancey PG, Zuo Y, Macrophage polarization by angiotensin II-type 1 receptor aggravates renal injury-acceleration of atherosclerosis: Arterioscler Thromb Vasc Biol, 2011; 31(12); 2856-64
61. Nainu F, Shiratsuchi A, Nakanishi Y, Induction of apoptosis and subsequent phagocytosis of virus-infected cells as an antiviral: Mech Front Immunol, 2017; 8; 1220
62. Xu Z, Shi L, Wang Y, Pathological findings of COVID-19 associated with acute respiratory distress syndrome: Lancet Respir Med, 2020; 8(4); 420-22
63. Chiu RW, Tang NL, Hui DS, ACE2 gene polymorphisms do not affect outcome of severe acute respiratory syndrome: Clin Chem, 2004; 50; 1683-86
64. Itoyama S, Keicho N, Hijikata M, Identification of an alternative 5′-untranslated exon and new polymorphisms of angiotensin-converting enzyme 2 gene: Lack of association with SARS in the Vietnamese population: Am J Med Genet A, 2005; 136; 52-57
65. Hussain M, Jabeen N, Raza F, Structural variations in human ACE2 may influence its binding with SARS-CoV-2 spike protein: J Med Virol, 2020 [Online ahead of print]
66. Karczewski KJ, Francioli LC, Tiao G, The mutational constraint spectrum quantified from variation in 141,456 humans: Nature, 2020; 581; 434-43
67. Zhao Y, Zhao Z, Wang Y, Single-cell RNA expression profiling of ACE2, the receptor of SARS-CoV-2: Am J Respir Crit Care Med, 2020; 202(5); 756-59
68. Xie X, Chen J, Wang X, Age- and gender-related difference of ACE2 expression in rat lung: Life Sci, 2006; 78(19); 2166-71
69. Cai G, Bossé Y, Xiao F, Tobacco smoking increases the lung gene expression of ACE2, the receptor of SARS-CoV-2: Am J Respir Crit Care Med, 2020; 201(12); 1557-59
70. Cai G: Tobacco-use disparity in gene expression of ACE2, the receptor of 2019-nCov, 2020 Preprints ()www.preprints.org
71. Smith JC, Sausville EL, Girish V, Cigarette smoke exposure and inflammatory signaling increase the expression of the SARS-CoV-2 receptor ACE2 in the respiratory tract: Dev Cell, 2020; 53(5); 514-29
72. Fu J, Zhou B, Zhang L, Expressions and significances of the angiotensin-converting enzyme 2 gene, the receptor of SARS-CoV-2 for COVID-19: Mol Biol Rep, 2020; 47(6); 4383-92
73. Samad A, Jafar T, Rafi JH, Identification of angiotensin-converting enzyme 2 (ACE2) protein as the potential biomarker in SARS-CoV-2 infection-related lung cancer using computational analysis: Genomics, 2020 [Online ahead of print]
74. Yu J, Ouyang W, Chua MLK, Xie C, SARS-CoV-2 transmission in patients with cancer at a tertiary care hospital in Wuhan, China: JAMA Oncol, 2020; 6(7); 1108-10
75. Menon J, Soto-Pantoja DR, Callahan MF, Angiotensin-(1–7) inhibits growth of human lung adenocarcinoma xenografts in nude mice through a reduction in cyclooxygenase-2: Cancer Res, 2007; 67; 2809-15
76. Qian YR, Guo Y, Wan HY, Angiotensin-converting enzyme 2 attenuates the metastasis of non-small cell lung cancer through inhibition of epithelial-mesenchymal transition: Oncol Rep, 2013; 29; 2408-14
77. Zhang Q, Lu S, Li T, ACE2 inhibits breast cancer angiogenesis via suppressing the VEGFa/VEGFR2/ERK pathway: J Exp Clin Cancer Res, 2019; 38(1); 173
78. Hulswit RJG, de Haan CAM, Bosch B-J, Coronavirus spike protein and tropism changes: Adv Virus Res, 2016; 96; 29-57
79. Bertram S, Glowacka I, Blazejewska P, TMPRSS2 and TMPRSS4 facilitate trypsin-independent spread of influenza virus in Caco-2 cells: J Virol, 2010; 84(19); 10016-25
80. Glowacka I, Bertram S, Müller MA, Evidence that TMPRSS2 activates the severe acute respiratory syndrome coronavirus spike protein for membrane fusion and reduces viral control by the humoral immune response: J Virol, 2011; 85(9); 4122-34
81. Stopsack KH, Mucci LA, Antonarakis ES, TMPRSS2 and COVID-19: Serendipity or opportunity for intervention?: Cancer Discov, 2020; 10(6); 779-82
Figures
Figure 1. Coronaviruses genome organization. Localized from the 5′ end, there are 2 open reading frames (ORF) – ORF1a and ORF1b – which encode polyproteins pp1a and pp1ab that are sequentially cleaved to release 16 or sometimes 15 functional nonstructural proteins constituting the replication-transcription complex (RTC). ORFs localized by the 3′ end encode structural and regulatory proteins: spike (S), ORF3A, envelope (E), membrane (M), ORF5, ORF6, ORF7A, ORF7B, ORF8, and nucleocapsid (N). The bottom panel shows the proteolytic processing of the pp1a and pp1ab polyproteins. The numbers of polyproteins pp1a and pp1ab segments correspond to nonstructural proteins: 3 – PLpro, papain-like proteinase; 5–3CLpro, 3C-like protease; 12 – RdRp, RNA-dependent RNA polymerase; 13 – HEL1, superfamily 1 helicase; 14 – ExoN, exoribonuclease, N7-MT, N7-methyl transferase; 15 – endoU, uridylate-specific endoribonuclease; and 16–2′-O-MT, 2′-O-methyl transferase. Functions of remaining nonstructural proteins are described in Table 1.Figure 2. Schematic diagram of alveolus during normal homeostasis (left panel) characterized by a balanced ACE1/ACE2 pathway, and under SARS-CoV-2 infection condition (right panel) where ACE2 receptor is downregulated by SARS-CoV-2 and increasing ACE1/ACE2 unbalance causes the renin-angiotensin system (RAS) hyper-activation leading to lung injury. AT I, AT II – alveolar type I, II epithelial cell (type I, II pneumocyte); ACE1, ACE2 – angiotensin-converting enzyme 1, 2; MasR – Mas receptor; Ang I, II – angiotensin I, II; AT1R, AT2R – angiotensin II type 1, 2 receptor. In Press
Clinical Research
Effects of Single-Bout Endurance Exercise Intensity on Peripheral Neurotrophic Factors in Patients With Isc...Med Sci Monit In Press; DOI: 10.12659/MSM.952089
Review article
Anisodus tanguticus in Cancer Research: A Review of Traditional Use, Phytochemistry, Extraction Methods, an...Med Sci Monit In Press; DOI: 10.12659/MSM.952999
Clinical Research
Nasal Mucociliary Clearance and Its Relationship With Disease Severity in Patients With Multiple SclerosisMed Sci Monit In Press; DOI: 10.12659/MSM.952850
Clinical Research
Modified Thoracoabdominal Nerves Block Through the Perichondrial Approach vs Subcostal Transversus Abdomini...Med Sci Monit In Press; DOI: 10.12659/MSM.953976
Most Viewed Current Articles
17 Jan 2024 : Review article 14,176,570
Vaccination Guidelines for Pregnant Women: Addressing COVID-19 and the Omicron VariantDOI :10.12659/MSM.942799
Med Sci Monit 2024; 30:e942799
13 Nov 2021 : Clinical Research 3,762,188
Acceptance of COVID-19 Vaccination and Its Associated Factors Among Cancer Patients Attending the Oncology ...DOI :10.12659/MSM.932788
Med Sci Monit 2021; 27:e932788
14 Dec 2022 : Clinical Research 2,466,310
Prevalence and Variability of Allergen-Specific Immunoglobulin E in Patients with Elevated Tryptase LevelsDOI :10.12659/MSM.937990
Med Sci Monit 2022; 28:e937990
16 May 2023 : Clinical Research 708,927
Electrophysiological Testing for an Auditory Processing Disorder and Reading Performance in 54 School Stude...DOI :10.12659/MSM.940387
Med Sci Monit 2023; 29:e940387