22 August 2022: Review Articles
Programmed Cell Death in Diabetic Nephropathy: A Review of Apoptosis, Autophagy, and Necroptosis
Nour S. Erekat1EF*DOI: 10.12659/MSM.937766
Med Sci Monit 2022; 28:e937766






Abstract
ABSTRACT: Diabetic nephropathy is a common complication of type I and type II diabetes, in which renal glomeruli are destroyed, resulting in renal damage, proteinuria, and hypertension. Apoptosis, autophagy, and necroptosis are 3 forms of programmed cell death that have been implicated in the pathogenesis of diabetic nephropathy. Apoptosis of podocytes leads to glomerular injury and podocyte depletion, which are associated with proteinuria and glomerular structural damage in diabetic nephropathy. Additionally, epithelial cells in the proximal convoluted tubules also undergo apoptosis in diabetic nephropathy, leading to tubular atrophy, which causes tubular cell depletion and the subsequent formation of atubular glomeruli in association with the loss of renal function. On the other hand, insufficiency of autophagy has been correlated with the pathogenesis of diabetic nephropathy. For instance, decreased autophagic activity has been shown in podocytes of the diabetic kidney, causing variations in podocyte function and subsequent disruption to the glomerular filtration barrier. Furthermore, attenuated autophagic activity has also been demonstrated in proximal tubular cells of the diabetic kidney, resulting in the buildup of impaired molecules and organelles, which are normally broken down by autophagy, leading to proteinuria. Moreover, necroptosis might have a key role in podocyte damage and subsequent decline in diabetic nephropathy. Thus, this article aims to review the mechanisms and effects of programmed cell death in diabetic nephropathy, including the roles of apoptosis, autophagy, and necroptosis.
Keywords: Apoptosis, Diabetic Nephropathies, review, Necroptosis, Hyperglycemia, Autophagy, Diabetes Mellitus, Type 2, Humans, Proteinuria
Background
Diabetes is a metabolic disorder that is characterized by hyperglycemia, which is high blood glucose level, as a consequence of deficiencies in insulin action, insulin secretion, or both [1]. Chronic hyperglycemia results in the dysfunction and failure of different organs, particularly the heart, blood vessels, eyes, and kidneys [2]. Diabetes can be classified into 3 types: type I, type II, and gestational diabetes [3]. Type I diabetes is insulin-dependent diabetes [4], whereas type II diabetes is non-insulin-dependent diabetes [5]. On the other hand, gestational diabetes is characterized by glucose intolerance that starts or that is recognized during pregnancy [3,6], and it increases the risk of development of type II diabetes [7,8]. Diabetes can have both microvascular and macrovascular complications, including nephropathy, cardiomyopathy, retinopathy, and peripheral neuropathy [1].
Diabetic nephropathy is a clinical syndrome, which is regarded as the leading cause of end-stage renal disease (ESRD) [9–11]. Diabetic nephropathy is characterized by albuminuria, which is increased albumin excretion in the urine [12]. Albuminuria is indicative of significantly increased cardiovascular mortality and morbidity in either type I or type II diabetes [13]. Albuminuria can be classified into 2 stages – macroalbuminuria and microalbuminuria – according to the amounts of albumin excreted in the urine [13]. Albumin excreted in the urine exceeds 200g/min in macroalbuminuria, and it is less than 199 g/min in microalbuminuria [13].
Diabetic nephropathy develops through several distinct phases [13]. Initially, functional alterations appear in the nephron at the level of the glomerulus, starting as glomerular hyperfiltration and hyperperfusion [14]. Then, the glomerular basement membrane thickens, and glomerular hypertrophy and sclerosis occur due to intraglomerular hypertension, and mesangial expansion that is caused by extracellular matrix accumulation [15].
The main risk factors for the progression of diabetic nephropathy comprise hyperglycemia, hypertension associated with obesity, and metabolic syndrome [13]. Diabetic nephropathy can be screened at the time of diagnosis in patients with type II diabetes by several methods, including measuring albumin-to-creatinine ratio in a random spot urine sample [16].
Apoptosis, autophagy, and necroptosis are 3 forms of programmed cell death that are implicated in the pathogenesis of diabetic nephropathy [17,18]. Apoptosis occurs via 2 well-characterized pathways, which are the extrinsic and intrinsic pathways [19]. Both apoptotic pathways are mediated by initiator and executioner caspases [20–22]. Apoptosis takes place in podocytes and epithelial cells of proximal convoluted tubules in association with the development of diabetic nephropathy [23,24].
Autophagy means self-eating and it is necessary for maintaining cell homeostasis under many stress circumstances [25]. Autophagy has been classified into macroautophagy, microautophagy, and chaperone-mediated autophagy [26]. Impaired autophagy in podocytes and proximal tubular cells has been correlated with the pathogenesis of diabetic nephropathy [27–29].
On the other hand, necroptosis is a programmed form of necrosis [30]. It might have a key role in podocyte damage and decline in diabetic nephropathy, particularly because it has been shown to be triggered subsequent to the inhibition of apoptosis that is induced by hyperglycemia [31].
Therefore, this article aims to review the mechanisms and effects of programmed cell death in diabetic nephropathy, including the roles of apoptosis, autophagy, and necroptosis.
Apoptosis
Apoptosis is also called type I programmed cell death [32]. It is morphologically characterized by cell shrinkage, chromatin condensation, nuclear deoxyribonucleic acid (DNA) fragmentation, and the formation of apoptotic bodies, which are eventually removed via phagocytosis, preventing the elicitation of any inflammation [33–35].
The initiation and execution of apoptosis are carried out by caspases, which are cysteine proteases that are specific for aspartate [36]. Caspases normally exist in the cytoplasm as inactive zymogens, which can be stimulated via cleavage [32,37]. Apoptotic caspases are functionally classified into initiator caspases (caspases-8, -9, and -10) and executioner caspases (caspases-3, -6, and -7) [38–40]. The initiator caspases are triggered by upstream signals, such as external cell death molecules and internal stress [20]. Initiator caspases are activated by autocleavage, and they subsequently stimulate the downstream executioner caspases [21,22].
There are 2 well-characterized apoptotic pathways (Figure 1), which are the extrinsic pathway (death receptor-mediated pathway) and the intrinsic pathway (mitochondria-mediated pathway) [19].
In the extrinsic pathway, the apoptotic signaling is initiated by the binding of extracellular ligands to the transmembrane domains of the extracellular death receptors [32]. Extracellular ligands include tumor necrosis factor (TNF) and TNF-related apoptosis-inducing ligand (TRAIL) that bind to type 1 TNF receptor (TNFR1), and TRAIL receptor, respectively [41]. Consequently, death receptors trimerize and engage adaptor molecules to their death domains, leading to the stimulation of caspase-8, which then activates the executioner caspases that mediate the proteolytic events of the apoptotic cell death, leading to destruction of the nucleus and other intracellular structures [41,42].
The intrinsic pathway is triggered by cellular stresses, such as DNA impairment and metabolic stress [43]. These stimuli cause an alteration in the equilibrium between pro-apoptotic B-cell lymphoma 2 (Bcl-2) family members, such as Bcl-2-associated X protein (Bax) and Brassinosteroid-insensitive 1 (BRI1)-associated kinase (Bak), and anti-apoptotic members, such as Bcl-2 and B-cell lymphoma-extra-large (Bcl-xL) [44]. Consequently, pro-apoptotic Bcl-2 family members accumulate on the outer membrane of mitochondria, resulting in mitochondrial outer membrane permeabilization (MOMP) [45]. Thus, cytochrome c is discharged from the mitochondrial intermembranous space to the cytoplasm [45]. Once in the cytoplasm, cytochrome c participates in generation of a multimeric apoptotic peptidase activating factor 1 (Apaf-1)/cytochrome c complex by binding to Apaf-1 in the presence of deoxyadenosine triphosphate (dATP) or adenosine triphosphate (ATP) [46]. Subsequently, the resultant complex recruits and activates caspase-9, leading to production of the apoptosome and consequent dissociation of active caspase-9, which cleaves and consequently stimulates the executioner caspases-3 and -7 [47,48].
Tumor suppressor protein (p53) is a transcription factor that is physiologically maintained at low levels [49–51]. P53 can stimulate apoptosis, principally depending on its transcriptional activity [52]. P53 can trigger the intrinsic apoptotic pathway by activating the transcription of many pro-apoptotic genes, such as the genes encoding Bax, phorbol-12-myristate-13-acetate-induced protein 1 (Noxa), and p53-upregulated modulator of apoptosis (Puma), which are pro-apoptotic homology (BH-3)-only Bcl-2 proteins [52]. Additionally, p53 stimulates the expression of Apaf-1, causing formation of the apoptosome [52]. On the other hand, p53 can repress anti-apoptotic genes, thus inducing apoptosis [52]. Moreover, p53 can trigger the extrinsic apoptotic pathway by activating the transcription of FS7-associated cell surface antigen (Fas) [52,53].
Autophagy
Autophagy is referred to as type II programmed cell death, and it means “self-eating” [54,55]. Normally, it happens at low basal levels, being essential for the clearance of impaired proteins and long-lived organelles [56]. Autophagy is essential for maintaining cell homeostasis under many stress circumstances [25]. Autophagy has been classified into 3 types, which vary in their mechanisms and functions [26]. Those 3 types are macroautophagy, microautophagy, and chaperone-mediated autophagy [26]. Macroautophagy is the predominant type, and it is denoted as “autophagy” [26].
Isolation membranes used in autophagy are assumed to derive from the endoplasmic reticulum (ER) membrane, and they lengthen and fuse, forming autophagosomes [57]. Autophagosomes engulf a portion of the cytoplasm [58]. Hence, the autophagic process occurs via 5 major stages (Figure 2), which are initiation, nucleation, elongation/closure, fusion, and breakdown [59]. Throughout these stages, autophagy-related genes (Atg) and proteins are implicated [60]. For instance, the mammalian ortholog of yeast Atg1 is called the unc51-like kinase 1 (Ulk1) complex, and it consists of Ulk1 serine/threonine protein kinase, Atg13, and focal adhesion kinase family interacting protein of 200 kD (FIP200), which is the mammalian homolog of yeast Atg17 [61,62]. Ulk1 can initiate autophagy by phosphorylating Atg13 and FIP200 [63]. Additionally, the mammalian ortholog of yeast Atg6 is called Beclin 1, and it is present with class III phosphoinositide 3-kinase (PI3K) in a complex product that is called phosphatidylinositol 3-phosphate (PtdIns(3)P) [64,65]. Beclin 1 is essential for phagophore nucleation [66]. Thus, autophagy is triggered by management with PtdIns(3)P, and it is prevented by blocking class III PI3K [67,68].
Once autophagy is initiated, a membrane sac isolation membrane extends around the substrate to be eliminated and eventually forms an autophagosome or autophagic vacuole [69]. Microtubule-associated protein light chain 3 (LC3) is the mammalian ortholog of yeast Atg8, and it is essential for autophagosome elongation/closure [70]. LC3 engaged on the surface of autophagosomes persists on the autophagosomal membrane [71]. It has been demonstrated that autophagosome formation needs 2 ubiquitin-like conjugation systems [72]. Subsequent to its formation, the autophagosome fuses with the lysosome [72]. Consequently, substrates enclosed by the autophagosome are broken down by hydrolases within the lysosomal cavity [72]. Protein p62, which is also called sequestosome 1, is continually hydrolyzed by autophagy-lysosome system after being recruited to the autophagosomes by LC3 interaction [73,74]. Thus, p62 buildup is detected in autophagy-deficient cells [74].
Autophagy is augmented by nutrient deprivation and extracellular or intracellular stress [75,76]. Autophagosome formation is intensely stimulated in response to starvation [77]. Nutrient status is sensed by 2 well-described signaling pathways, which are the pathways of adenosine monophosphate-activated protein kinase (AMPK) and mammalian target of rapamycin complex 1 (mTORC1) [78].
Autophagy is activated by AMPK, which is a critical energy sensor of adenosine monophosphate (AMP), and it is increased by a rise in intracellular levels of AMP [78]. AMPK senses the energy state of a cell by detecting the AMP/ATP ratio [79]. For example, AMPK is stimulated in low glucose conditions, which lead to decreased levels of ATP and activation of autophagy [80,81]. AMPK can be activated by several kinases, such as liver kinase B1, calcium/calmodulin kinase, and transforming growth factor β-activated kinase 1 (TAK1) [82]. AMPK can trigger autophagy by reducing mTORC1 activity and/or regulating Ulk1 phosphorylation directly [83,84]. mTOR is a protein kinase that makes up 2 functional complexes, which are known as mTORC1 and mTORC2 [85]. mTORC1 is a rapamycin-sensitive protein kinase complex that controls various cellular processes, such as autophagy, based on the cellular nutritional status [85]. mTORC1 activity is definitely controlled by Ras homology enriched in brain (Rheb), which is a GTP-binding protein [85–87]. Protein kinase B (Akt) is phosphorylated by insulin signal via PI3K and phosphoinositide-dependent kinase-1, and it consequently attenuates tuberous sclerosis 2, which is a potent Rheb suppressor [88,89]. Thus, an insulin signal reduces autophagy through activating mTORC1 in cells [90]. Activation of mTORC1 also needs amino acids, and it is associated with the movement of mTORC1 from the cytoplasm to lysosomal membranes [91]. Once activated, mTORC1 phosphorylates Ulk1 inhibiting it and subsequently preventing autophagosome formation [85].
Autophagy can also be triggered by reactive oxygen species (ROS) [92]. For instance, exogenous hydrogen peroxide can eventually activate Atg4 proteases, resulting in upregulation of proteolytic mature LC3 and the suppression of mTORC1 activity [93]. Consequent to increased ROS, mitogen-activated protein kinases, such as c-Jun N-terminal kinase 1 (JNK1), are often activated, leading to activation of autophagy [94]. Thus, autophagy of damaged mitochondria, which is called mitophagy, is essential to inhibit ROS buildup [95]. Additionally, autophagy is induced by hypoxia via activating hypoxia-inducible factor 1 (HIF1) transcription factor, which ultimately leads to release of Beclin-1, which triggers autophagy [96].
Necroptosis
Necroptosis is a type of programmed cell death that has morphological features similar to those of necrosis, including swollen organelles and fragmented nuclear membrane [30]. Additionally, the integrity of the plasma membrane is impaired, leading to its rupture and the subsequent leakage of cellular substances, eventually resulting in inflammation [97].
Necroptosis is triggered by interleukin-1beta (IL-1β), TNF, certain viral infections, and other factors [98]. Furthermore, necroptosis depends on receptor-interacting serine/threonine kinase 1 (RIPK1) to RIPK3, which form an RIPK1/RIPK3 complex, which is known as the necrosome, which activates the necroptotic pathway in the absence of caspase-8 [98]. Thus, RIPK1 and RIPK3 are phosphorylated, leading to the recruitment and activation of pseudokinase [98]. Consequently, plasma membrane integrity is impaired and subsequently ruptured, leading to discharge of damage-associated molecular patterns (DAMPs) [97]. DAMPs, which comprise IL-1 family members, stimulate the inflammasome, activate inflammation, and provoke immune reactions [97]. RIPK3-dependent necroptosis is regulated by the ubiquitin-proteasome system [97]. Thus, RIPK1 and RIPK3 are activated by deubiquitinating enzymes, stimulating necroptosis [97,99].
Apoptosis in Diabetic Nephropathy
Hyperglycemia and insulin resistance are implicated in the pathogenesis of diabetic nephropathy [9,100–102]. Hyperglycemia is believed to cause microvascular injury, particularly in the renal glomerulus, leading to development of diabetic nephropathy [103]. Hyperglycemia induces reactive oxygen species generation, which initiates podocyte apoptosis (Figure 3) and subsequent podocyte decline, both in vitro and in vivo [104]. Apoptosis leads to glomerular injury in diabetic kidneys, decreasing podocyte numbers [23]. The decreased number of podocytes is associated with proteinuria and glomerular structural damage in diabetic nephropathy [105,106]. Podocyte apoptosis and the subsequent podocyte depletion may indeed represent early events affecting the diabetic kidney and underlying diabetic glomerulopathy, which leads to diabetic nephropathy in type I and type II diabetes [107,108]. Podocyte apoptosis coincides with the onset of diabetes and hyperglycemia in type I and type II diabetes, indicating that glucotoxicity may be underlying the stimulation of pro-apoptotic signaling and subsequent podocyte injury in diabetic kidneys. Decreased glomerular podocyte density precedes microalbuminuria [109].
P53 mediates podocyte apoptosis in cells exposed to high glucose [110,111]. Indeed, high glucose increases p53 messenger ribonucleic acid (mRNA) and protein expression (Figure 3), which activates Bax and the intrinsic apoptotic pathway, with the eventual activation of caspase-3 in podocytes, leading to their apoptosis [110,111]. P53 downregulation blocks podocyte apoptosis that is induced by high glucose [112].
Epithelial cells in the proximal convoluted tubules also undergo apoptosis in diabetic nephropathy, leading to tubular atrophy [24]. Thus, diabetic nephropathy is characterized by the absence of a connection between glomeruli and proximal convoluted tubules due to tubular atrophy, which is associated with loss of renal function [113]. Tubular atrophy leads to tubular cell depletion and the subsequent formation of atubular glomeruli [114].
Hyperglycemia modulates the expression of apoptosis regulatory genes in renal proximal tubular cells, leading to their cell death [115]. Hyperglycemia induces angiotensinogen gene expression through reactive oxygen species formation in the proximal convoluted tubules [115]. Renin-angiotensin has been suggested to be involved in tubular epithelial cell apoptosis in diabetic kidneys (Figure 3), since blockade of renin-angiotensin system has been demonstrated to attenuate the apoptosis of tubular epithelial cells [115]. Additionally, hyperglycemia is associated with increased reactive oxygen species generation, which induces apoptosis in proximal convoluted tubules, leading to tubular atrophy and subsequent formation of atubular glomeruli [116–118].
Tubular apoptosis has been reported to be associated with glycogen accumulation, as well as other factors of the diabetic condition [119]. Glycogen-accumulating cells undergo apoptosis, leading to renal tubular atrophy, which contributes to depletion of renal tubular epithelial cells, representing a hallmark feature of end-stage diabetic nephropathy [119].
Autophagy in Diabetic Nephropathy
Hyperglycemia inhibits autophagy, because it causes overproduction of irreversibly glycated proteins, which are called advanced glycation end-products (AGEs) [120]. Consistently, AGEs have been demonstrated to inhibit autophagy in cultured proximal tubular epithelial cells (PTECs) [121]. Thus, AGE degradation, which takes place in PTECs, is impaired, causing their accumulation and subsequent inflammation, which is a risk factor for development of diabetic nephropathy [121]. Consequently, AGE surplus disturbs the permeabilization of lysosomal membrane and consequently impairs autophagy in diabetic nephropathy, causing tubular injury, inflammation, and interstitial fibrosis [121].
Diabetic nephropathy is characterized by proteinuria, which results from cell death of podocytes and dysfunction of their foot processes, leading to destruction of the glomerular filtration barrier [122]. Podocytes are highly specialized glomerular epithelial cells that enable the glomerular filtration barrier (GFB) to perform its function by wrapping around the outer aspect of the glomerular basement membrane (GBM) with their interdigitating foot processes [122]. However, podocyte loss frequently occurs in diabetic nephropathy, leading to proteinuria [122]. Podocytes have constitutively high basal autophagic activity under normal conditions, suggesting the necessity of autophagy for maintaining podocyte homeostasis [123]. Accordingly, conserving podocyte cell homeostasis is considered as a therapeutic approach to inhibit the progression of diabetic nephropathy to nephrotic syndrome [124]. Indeed, impaired autophagy causes podocyte loss [125]. For instance, deleting Atg5 gene in podocytes leads to glomerular lesions that are concomitant with podocyte loss and albuminuria [126].
Autophagy is speculated to be involved in the pathogenesis of diabetic nephropathy [127]. Decreased autophagic activity has been demonstrated in podocytes after induction of diabetes using streptozotocin [27]. Consistently, hyperglycemia has been suggested to decrease autophagic activity (Figure 3), causing variations in podocyte function and subsequent disruption of the glomerular filtration barrier [27]. For example, cultured podocytes exhibited decreased autophagic activity and associated proteins, including Beclin-1 and the Atg5-Atg12 complex, consequent to their exposure to high concentrations of glucose [27,28]. Likewise, an in vitro study showed that underexpression of Beclin-1 in podocytes eventually caused albumin leakage [128]. Additionally, morphologically abnormal lysosomes were substantially increased subsequent to autophagy deficiency in the podocytes of obese type II diabetic rodents [129,130]. Thus, impaired lysosomes are proposed to be a significant degradation target of autophagy in podocytes under diabetic circumstances [130].
Unlike podocytes, proximal tubular cells exhibit very low basal levels of autophagy under normal conditions [131]. However, autophagy in proximal tubular cells is increased in stress conditions, such as those caused by acute kidney injury consequent to ischemia and nephrotoxic agents, such as cisplatin [132–134]. Additionally, activation of autophagy in proximal tubular cells was suggested to be renoprotective to manage with both acute and chronic nephrotoxic stresses [134]. Deficiency of autophagy in proximal tubular cells after deletion of Atg5 gene led to proteinuria and subsequent tubulointerstitial lesions [133]. Similar to other proteinuric kidney diseases, proteinuria causes strong nephrotoxic stress and activates autophagy in diabetic nephropathy [124].
On the other hand, induction of diabetes has been demonstrated to attenuate autophagic activity in proximal tubular cells [29]. Indeed, autophagy has been attenuated in kidneys after induction of diabetes using streptozotocin in mice, resulting in buildup of impaired molecules and organelles that are normally broken down by autophagy [29,124]. Additionally, accumulation of damaged molecules indicative of reduced autophagy has been found in biopsies from type II diabetic patients [124]. Furthermore, mTORC1 has been implicated in prevention of autophagy associated with diabetes in proximal tubular cells in type II diabetic humans and mice, indicating an intimate correlation between insufficient autophagy and the pathogenesis of diabetic nephropathy [135]. Thus, enhancing autophagy may be regarded as a potential therapeutic approach for diabetic nephropathy [124].
Necroptosis in Diabetic Nephropathy
Necroptosis is involved in the pathogenesis of diabetic nephropathy [136]. Podocyte necroptosis has been shown to be stimulated by ubiquitin C-terminal hydrolase L1 (UCHL1), which regulates the ubiquitination state of the RIPK1/RIPK3 pathway [31,137]. Indeed, abnormal overexpression of UCHL1 in podocytes stimulates podocyte injury, leading to polyubiquitin buildup [31]. Therefore, upregulation of UCHL1 in diabetic nephropathy has been associated with the dysregulation of ubiquitination of the RIPK1/RIPK3 pathway, resulting in necroptosis of podocytes [31]. It has been shown that inhibition of apoptosis induced by hyperglycemia could trigger necroptosis [31]. Thus, necroptosis might have a key role in podocyte damage and subsequent decline in diabetic nephropathy via activating the RIPK1-RIPK3 signaling pathway (Figure 3) [31]. Finally, necroptosis was reported to be obstructed by necrostatin-1 (Nec-1), which blocks the RIPK1/RIPK3 pathway, and genetic deletion of UCHL1 decreased the expression of RIPK1 and RIPK3 [31].
Therapeutic Implications of Programmed Cell Death in Diabetic Nephropathy
Because apoptosis, autophagy, and/or necroptosis are involved in diabetic nephropathy, therapeutic strategies that impede the molecular and biochemical steps involved in one or more types of programmed cell death that occur in diabetic nephropathy could be renoprotective (Table 1).
Since apoptosis is mediated by p53, p53 downregulation has been proposed as a possible anti-apoptotic therapeutic method in diabetic nephropathy that develops via apoptosis [111,138,139]. Additionally, management with caspase inhibitors has been reported to impede the development of diabetic nephropathy, halting cell death [140]. Drugs, such as paricalcitol and/or enalapril, which improve the antioxidant defense system attenuating oxidative stress, have been reported to exert renoprotective effects in streptozotocin-induced diabetic renopathy by suppressing apoptosis via reducing the renal expression of pro-apoptotic p53 and caspase-3 and augmenting the expression of the anti-apoptotic Bcl-2 [140]. Additionally, catalase overexpression exerts beneficial effects, attenuating apoptosis and pro-apoptotic gene expression in the proximal convoluted tubules [141–143]. Furthermore, taurine, which is a conditionally essential amino acid, has been shown to act as an endogenous antioxidant in renal tubular cells, attenuating hyperglycemia-induced apoptosis in human tubular cells by preventing oxidative stress [144]. Thus, the inhibition of oxidative stress by taurine has been demonstrated to prevent tubulointerstitial injury in diabetic nephropathy [144].
Moreover, long intergenic noncoding RNAs (lincRNAs) are involved in the pathogenesis of diabetic nephropathy [145]. However, miR-27a-3p inhibition, which is one of the microRNAs (miRNAs) that prevent gene expression post-transcriptionally, has been shown to slow the progression of diabetic nephropathy by suppressing podocyte apoptosis via upregulating tissue inhibitor of matrix metalloproteinases-3 (TIMP3) [145].
Autophagy in proximal tubular cells is triggered by temporary food deprivation. Additionally, calorie restriction exerts beneficial effects in several kinds of renal injury [146]. Thus, a calorie restriction regimen must be regarded as an effective therapeutic approach to inhibit diabetic nephropathy. Calorie restriction enhances renal impairment in type II diabetic Wistar fatty rats, and it repairs autophagy activity in their proximal tubular cells [147]. Therefore, an agent that can imitate caloric restriction might be a potential therapy for diabetic nephropathy by inducing autophagy in mammalian cells. Moreover, rapamycin, which induces autophagy by inhibiting mTORC1, has been reported to decrease glomerular lesions in experimental diabetic nephropathy [124,148]. Thus, rapamycin might be protective in diabetic nephropathy by activating autophagy. However, extreme mTORC1 prevention impairs podocyte function, suggesting that further investigation is required regarding the safety and effectiveness of mTORC1 prevention in patients with diabetic nephropathy [147,148].
Deactivated AMPK may be involved in the development of diabetic nephropathy by suppressing autophagy, suggesting that AMPK stimulation might be targeted for repairing autophagy in diabetic kidneys [149]. Indeed, AMPK deactivation is inverted by agents such as metformin and resveratrol, which also reduce diabetic glomerular and tubular lesions [150–153]. AMPK activation can induce autophagy, which may mediate its renoprotective mechanism, sustaining renal homeostasis in diabetic kidneys [151,153]. Glomeruli and tubules of both type I and type II diabetic animal models display dephosphorylation that deactivates AMPK [154,155].
Finally, inhibition of necroptosis might be a possible therapeutic method in diabetic nephropathy, since it has been demonstrated to improve diabetic nephropathy [156]. For instance, adiponectin could reduce podocyte necroptosis and subsequent damage in diabetic nephropathy by reducing the expression levels of RIPK1 and RIPK3 [156]. In addition, necrostatin-1 (Nec1) inhibited necroptosis and the associated expression of RIPK1 and RIPK3, which were stimulated subsequent to apoptosis inhibition using caspase inhibitors in podocytes exposed to high glucose [31]. Additionally, genetic deletion of UCHL1 resulted in decreased half-life of RIPK1 and RIPK3 proteins and underexpression of RIPK1 and RIPK3 [31]. Therefore, UCHL1 inhibition might be a potential therapeutic strategy to suppress the necroptotic RIPK1/RIPK3 pathway in an attempt to rescue podocytes in patients with diabetic nephropathy [31].
Conclusions
There is ample evidence supporting the involvement of programmed cell death, including apoptosis, autophagy, and necroptosis, in the pathogenesis of diabetic nephropathy. A better comprehension of the molecular events regulating apoptosis, autophagy, and necroptosis, which are associated with the pathogenesis of diabetic nephropathy, would be tremendously helpful in discovering factors that can serve as potential therapeutic targets to stop or retard the progression of the diabetic nephropathy. Additionally, further investigation in this field can be predicted to afford promising therapeutic approaches, which might decrease the incidence of diabetic nephropathy development in diabetic patients via manipulating cell death programs.
Figures
![Extrinsic and intrinsic pathways of apoptosis. The extrinsic apoptotic pathway is triggered by the binding of a particular death ligand to death receptor, leading to its trimerization and the consequent recruitment of procaspase-8, which is subsequently activated. In contrast, the intrinsic apoptotic pathway starts in response to apoptotic stimuli that induce pro-apoptotic proteins, such as B-cell lymphoma 2-associated X protein (Bax), triggering the permeabilization of the outer mitochondrial membrane, and the consequent release of cytochrome c into the cytosol, where it contributes to the formation of a multimeric apoptotic peptidase activating factor 1 (Apaf-1)/cytochrome c complex that recruits procaspase-9, forming the apoptosome. Hence, procaspase-9 is activated and consequently detached from the complex. Ultimately, active caspase-8 and active caspase-9 activate executioner caspases-3, -6, and/or -7, which mediate apoptosis. This figure was adapted with permission from John Wiley and Sons, from “Apoptosis and its therapeutic implications in neurodegenerative diseases” by Nour S. Erekat, 2022, Clinical Anatomy [34].](https://jours.isi-science.com/imageXml.php?i=medscimonit-28-e937766-g001.jpg&idArt=937766&w=1000) Figure 1. Extrinsic and intrinsic pathways of apoptosis. The extrinsic apoptotic pathway is triggered by the binding of a particular death ligand to death receptor, leading to its trimerization and the consequent recruitment of procaspase-8, which is subsequently activated. In contrast, the intrinsic apoptotic pathway starts in response to apoptotic stimuli that induce pro-apoptotic proteins, such as B-cell lymphoma 2-associated X protein (Bax), triggering the permeabilization of the outer mitochondrial membrane, and the consequent release of cytochrome c into the cytosol, where it contributes to the formation of a multimeric apoptotic peptidase activating factor 1 (Apaf-1)/cytochrome c complex that recruits procaspase-9, forming the apoptosome. Hence, procaspase-9 is activated and consequently detached from the complex. Ultimately, active caspase-8 and active caspase-9 activate executioner caspases-3, -6, and/or -7, which mediate apoptosis. This figure was adapted with permission from John Wiley and Sons, from “Apoptosis and its therapeutic implications in neurodegenerative diseases” by Nour S. Erekat, 2022, Clinical Anatomy [34].
Figure 1. Extrinsic and intrinsic pathways of apoptosis. The extrinsic apoptotic pathway is triggered by the binding of a particular death ligand to death receptor, leading to its trimerization and the consequent recruitment of procaspase-8, which is subsequently activated. In contrast, the intrinsic apoptotic pathway starts in response to apoptotic stimuli that induce pro-apoptotic proteins, such as B-cell lymphoma 2-associated X protein (Bax), triggering the permeabilization of the outer mitochondrial membrane, and the consequent release of cytochrome c into the cytosol, where it contributes to the formation of a multimeric apoptotic peptidase activating factor 1 (Apaf-1)/cytochrome c complex that recruits procaspase-9, forming the apoptosome. Hence, procaspase-9 is activated and consequently detached from the complex. Ultimately, active caspase-8 and active caspase-9 activate executioner caspases-3, -6, and/or -7, which mediate apoptosis. This figure was adapted with permission from John Wiley and Sons, from “Apoptosis and its therapeutic implications in neurodegenerative diseases” by Nour S. Erekat, 2022, Clinical Anatomy [34]. 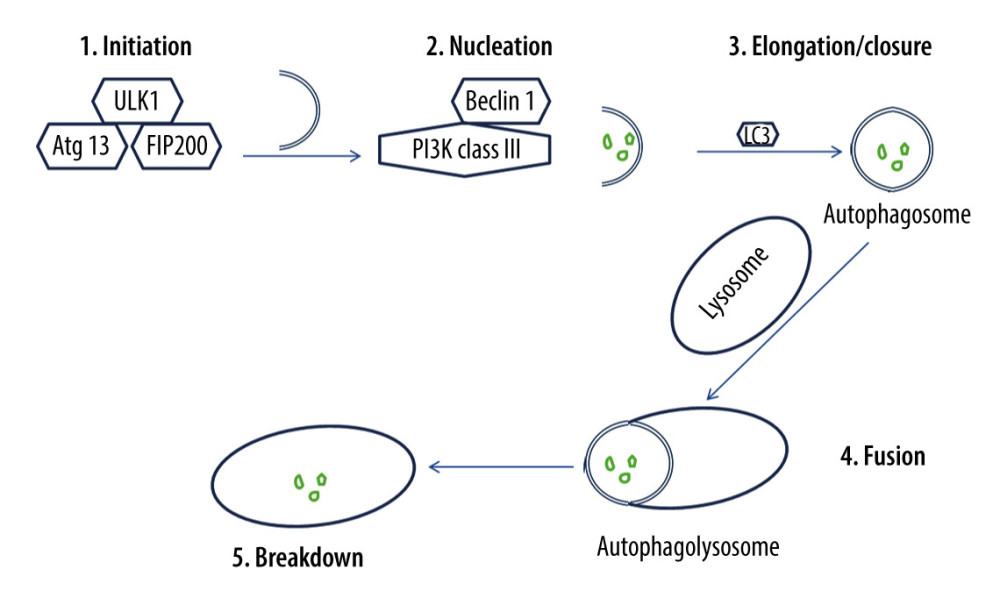 Figure 2. The process of autophagy. The autophagic process occurs via 5 major stages, which are initiation, nucleation, elongation/closure, fusion, and breakdown. Initiation stage: The unc51-like kinase 1 (Ulk1) complex consists of Ulk1 serine/threonine protein kinase, autophagy-related gene 13 (Atg13), and focal adhesion kinase family interacting protein of 200 kD (FIP200). Ulk1 is required for the initiation stage. 2. Nucleation stage: Beclin 1, which exists with class III phosphoinositide 3-kinase (PI3K), is essential for the nucleation stage. 3. Elongation/closure stage: A membrane sac isolation membrane wraps around the degradation substrates and ultimately closes to become an autophagosome. This stage requires the microtubule-associated protein light chain 3 (LC3). 4. Fusion stage: the outer membrane of the autophagosome completely fuses with the outer lysosomal membrane. 5. Breakdown stage: contents of the autophagosome are released and subsequently degraded into the lysosomal cavity. This figure was generated using Microsoft PowerPoint Software, version 10, Microsoft Corp., USA.
Figure 2. The process of autophagy. The autophagic process occurs via 5 major stages, which are initiation, nucleation, elongation/closure, fusion, and breakdown. Initiation stage: The unc51-like kinase 1 (Ulk1) complex consists of Ulk1 serine/threonine protein kinase, autophagy-related gene 13 (Atg13), and focal adhesion kinase family interacting protein of 200 kD (FIP200). Ulk1 is required for the initiation stage. 2. Nucleation stage: Beclin 1, which exists with class III phosphoinositide 3-kinase (PI3K), is essential for the nucleation stage. 3. Elongation/closure stage: A membrane sac isolation membrane wraps around the degradation substrates and ultimately closes to become an autophagosome. This stage requires the microtubule-associated protein light chain 3 (LC3). 4. Fusion stage: the outer membrane of the autophagosome completely fuses with the outer lysosomal membrane. 5. Breakdown stage: contents of the autophagosome are released and subsequently degraded into the lysosomal cavity. This figure was generated using Microsoft PowerPoint Software, version 10, Microsoft Corp., USA. 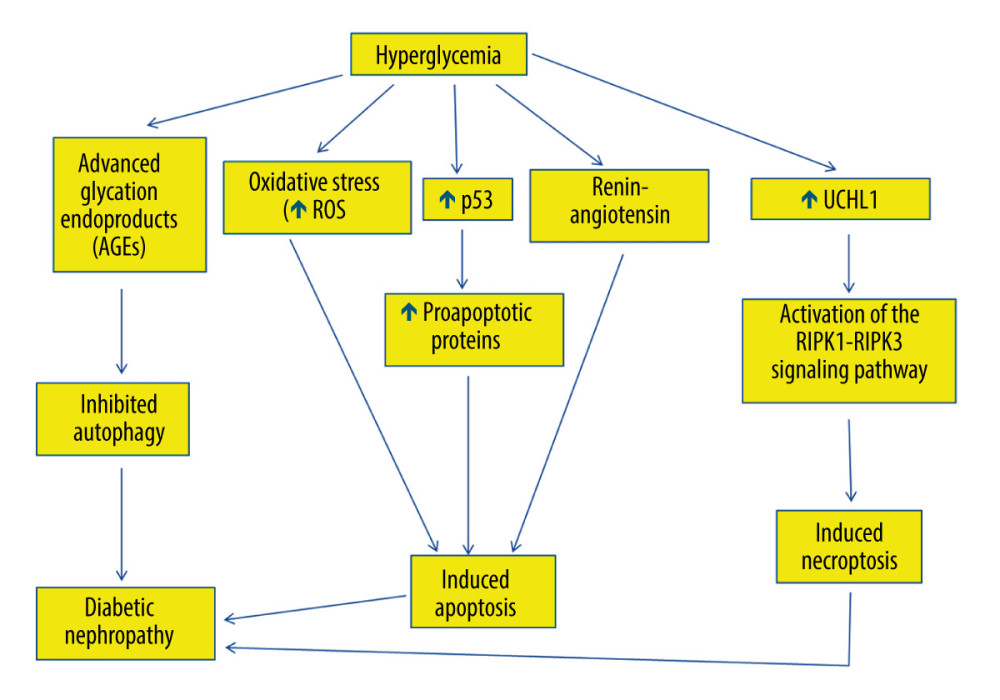 Figure 3. The role of hyperglycemia in the suppression of autophagy and induction of apoptosis and necroptosis in diabetic nephropathy. Hyperglycemia causes overproduction of advanced glycation end-products (AGEs), which inhibit autophagy, leading to diabetic nephropathy. Hyperglycemia induces reactive oxygen species generation, which initiates apoptosis. Hyperglycemia also increases tumor suppressor p53 messenger ribonucleic acid (mRNA) and protein expression, thus activating the transcription of many pro-apoptotic proteins that stimulate the extrinsic and the intrinsic apoptotic pathways. Additionally, hyperglycemia stimulates renin-angiotensin, which is involved in apoptosis and is involved in diabetic nephropathy. Hyperglycemia can also trigger necroptosis in diabetic nephropathy via activating the interacting serine/threonine kinase 1 (RIPK1)- RIPK3 signaling pathway. This figure was generated using Microsoft PowerPoint Software, version 10, Microsoft Corp., USA.
Figure 3. The role of hyperglycemia in the suppression of autophagy and induction of apoptosis and necroptosis in diabetic nephropathy. Hyperglycemia causes overproduction of advanced glycation end-products (AGEs), which inhibit autophagy, leading to diabetic nephropathy. Hyperglycemia induces reactive oxygen species generation, which initiates apoptosis. Hyperglycemia also increases tumor suppressor p53 messenger ribonucleic acid (mRNA) and protein expression, thus activating the transcription of many pro-apoptotic proteins that stimulate the extrinsic and the intrinsic apoptotic pathways. Additionally, hyperglycemia stimulates renin-angiotensin, which is involved in apoptosis and is involved in diabetic nephropathy. Hyperglycemia can also trigger necroptosis in diabetic nephropathy via activating the interacting serine/threonine kinase 1 (RIPK1)- RIPK3 signaling pathway. This figure was generated using Microsoft PowerPoint Software, version 10, Microsoft Corp., USA. 
References
1. Zaccardi F, Webb DR, Yates T, Davies MJ, Pathophysiology of type 1 and type 2 diabetes mellitus: A 90-year perspective: Postgrad Med J, 2016; 92(1084); 63-69
2. Giri B, Dey S, Das T, Chronic hyperglycemia mediated physiological alteration and metabolic distortion leads to organ dysfunction, infection, cancer progression and other pathophysiological consequences: An update on glucose toxicity: Biomed Pharmacother, 2018; 107; 306-28
3. Khan RMM, Chua ZJY, Tan JC, From pre-diabetes to diabetes: Diagnosis, treatments and translational research: Medicina (Kaunas), 2019; 55(9); 546
4. Schmidt AM, Highlighting diabetes mellitus: The epidemic continues: Arterioscler Thromb Vasc Biol, 2018; 38(1); e1-e8
5. Weber P, Weberova D, Meluzinova H, How to approach to the therapy of diabetes in the elderly: Adv Gerontol, 2014; 27(3); 519-30
6. Lende M, Rijhsinghani A, Gestational diabetes: Overview with emphasis on medical management: Int J Environ Res Public Health, 2020; 17(24); 9573
7. Zhu Y, Zhang C, Prevalence of gestational diabetes and risk of progression to type 2 diabetes: A global perspective: Curr Diab Rep, 2016; 16(1); 7
8. Vounzoulaki E, Khunti K, Abner SC, Progression to type 2 diabetes in women with a known history of gestational diabetes: Systematic review and meta-analysis: BMJ, 2020; 369; m1361
9. Samsu N, Diabetic nephropathy: Challenges in pathogenesis, diagnosis, and treatment: Biomed Res Int, 2021; 2021; 1497449
10. Doshi SM, Friedman AN, Diagnosis and management of type 2 diabetic kidney disease: Clin J Am Soc Nephrol, 2017; 12(8); 1366-73
11. Podgorski P, Konieczny A, Lis L, Glomerular podocytes in diabetic renal disease: Adv Clin Exp Med, 2019; 28(12); 1711-15
12. Sagoo MK, Gnudi L, Diabetic nephropathy: An overview: Methods Mol Biol, 2020; 2067; 3-7
13. Lin YC, Chang YH, Yang SY, Update of pathophysiology and management of diabetic kidney disease: J Formos Med Assoc, 2018; 117(8); 662-75
14. Tonneijck L, Muskiet MH, Smits MM, Glomerular hyperfiltration in diabetes: Mechanisms, clinical significance, and treatment: J Am Soc Nephrol, 2017; 28(4); 1023-39
15. Chagnac A, Zingerman B, Rozen-Zvi B, Herman-Edelstein M, Consequences of glomerular hyperfiltration: The role of physical forces in the pathogenesis of chronic kidney disease in diabetes and obesity: Nephron, 2019; 143(1); 38-42
16. Lin CW, Mostafa NM, Andress DL, Relationship between atrasentan concentrations and urinary albumin to creatinine ratio in western and japanese patients with diabetic nephropathy: Clin Ther, 2018; 40(2); 242-51
17. Ding Y, Choi ME, Autophagy in diabetic nephropathy: J Endocrinol, 2015; 224(1); R15-30
18. Turkmen K, Inflammation, oxidative stress, apoptosis, and autophagy in diabetes mellitus and diabetic kidney disease: The Four Horsemen of the Apocalypse: Int Urol Nephrol, 2017; 49(5); 837-44
19. Tummers B, Green DR, Caspase-8: Regulating life and death: Immunol Rev, 2017; 277(1); 76-89
20. Prokhorova EA, Kopeina GS, Lavrik IN, Zhivotovsky B, Apoptosis regulation by subcellular relocation of caspases: Sci Rep, 2018; 8(1); 12199
21. Wang Y, Hu S, Tuerdi M, Initiator and executioner caspases in salivary gland apoptosis of Rhipicephalus haemaphysaloides: Parasit Vectors, 2020; 13(1); 288
22. Anson F, Thayumanavan S, Hardy JA, Exogenous introduction of initiator and executioner caspases results in different apoptotic outcomes: JACS Au, 2021; 1(8); 1240-56
23. Jin J, Shi Y, Gong J, Exosome secreted from adipose-derived stem cells attenuates diabetic nephropathy by promoting autophagy flux and inhibiting apoptosis in podocyte: Stem Cell Res Ther, 2019; 10(1); 95
24. Kostic S, Hauke T, Ghahramani N, Expression pattern of apoptosis-inducing factor in the kidneys of streptozotocin-induced diabetic rats: Acta Histochem, 2020; 122(8); 151655
25. Catalani E, Giovarelli M, Zecchini S, Oxidative stress and autophagy as key targets in melanoma cell fate: Cancers (Basel), 2021; 13(22); 5791
26. Yan S, Huda N, Khambu B, Yin XM, Relevance of autophagy to fatty liver diseases and potential therapeutic applications: Amino Acids, 2017; 49(12); 1965-79
27. Guo H, Wang Y, Zhang X, Astragaloside IV protects against podocyte injury via SERCA2-dependent ER stress reduction and AMPKalpha-regulated autophagy induction in streptozotocin-induced diabetic nephropathy: Sci Rep, 2017; 7(1); 6852
28. Audzeyenka I, Rogacka D, Piwkowska A, Viability of primary cultured podocytes is associated with extracellular high glucose-dependent autophagy downregulation: Mol Cell Biochem, 2017; 430(1–2); 11-19
29. Lee YH, Kim SH, Kang JM, Empagliflozin attenuates diabetic tubulopathy by improving mitochondrial fragmentation and autophagy: Am J Physiol Renal Physiol, 2019; 317(4); F767-80
30. Liu C, Zhang K, Shen H, Necroptosis: A novel manner of cell death, associated with stroke (review): Int J Mol Med, 2018; 41(2); 624-30
31. Xu Y, Gao H, Hu Y, High glucose-induced apoptosis and necroptosis in podocytes is regulated by UCHL1 via RIPK1/RIPK3 pathway: Exp Cell Res, 2019; 382(2); 111463
32. Erekat NS, Apoptosis and its role in Parkinson’s disease: Parkinson’s disease: Pathogenesis and clinical aspects, 2018, Brisbane (AU)
33. Erekat NS, Cerebellar Purkinje cells die by apoptosis in the shaker mutant rat: Brain Res, 2017; 1657; 323-32
34. Erekat NS, Apoptosis and its therapeutic implications in neurodegenerative diseases: Clin Anat, 2022; 35(1); 65-78
35. Erekat NS, Programmed cell death in cerebellar Purkinje neurons: J Integr Neurosci, 2022; 21(1); 30
36. Wang X, Li F, Liu J, New insights into the mechanism of hepatocyte apoptosis induced by typical organophosphate ester: An integrated in vitro and in silico approach: Ecotoxicol Environ Saf, 2021; 219; 112342
37. Ivanisenko NV, Lavrik INMechanisms of procaspase-8 activation in the extrinsic programmed cell death pathway: Mol Biol (Mosk), 2019; 53(5); 830-37
38. Vogeler S, Carboni S, Li X, Joyce A, Phylogenetic analysis of the caspase family in bivalves: Implications for programmed cell death, immune response and development: BMC Genomics, 2021; 22(1); 80
39. Erekat NS, Al-Jarrah MD, Association of Parkinson disease induction with cardiac upregulation of apoptotic mediators P53 and active caspase-3: An immunohistochemistry study: Med Sci Monit Basic Res, 2018; 24; 120-26
40. Al-Jarrah MD, Erekat NS, Endurance exercise training suppresses Parkinson disease-induced overexpression of apoptotic mediators in the heart: NeuroRehabilitation, 2021; 48(3); 315-20
41. Mandal R, Barron JC, Kostova I, Caspase-8: The double-edged sword: Biochim Biophys Acta Rev Cancer, 2020; 1873(2); 188357
42. Erekat NS, Cerebellar upregulation of cell surface death receptor-mediated apoptotic factors in harmaline-induced tremor: An immunohistochemistry study: J Cell Death, 2018; 11; 1179066018809091
43. Ding Y, Wang H, Niu J, Induction of ROS overload by alantolactone prompts oxidative DNA damage and apoptosis in colorectal cancer cells: Int J Mol Sci, 2016; 17(4); 558
44. Soini T, Pihlajoki M, Kyronlahti A, Downregulation of transcription factor GATA4 sensitizes human hepatoblastoma cells to doxorubicin-induced apoptosis: Tumour Biol, 2017; 39(3); 1010428317695016
45. Morris DL, Tjandra N, Inducible fold-switching as a mechanism to fibrillate pro-apoptotic BCL-2 proteins: Biopolymers, 2021; 112(10); e23424
46. Elena-Real CA, Diaz-Quintana A, Gonzalez-Arzola K, Cytochrome c speeds up caspase cascade activation by blocking 14-3-3epsilon-dependent Apaf-1 inhibition: Cell Death Dis, 2018; 9(3); 365
47. Dorstyn L, Akey CW, Kumar S, New insights into apoptosome structure and function: Cell Death Differ, 2018; 25(7); 1194-208
48. Erekat NS, Active caspase-3 upregulation is augmented in at-risk cerebellar Purkinje cells following inferior olive chemoablation in the shaker mutant rat: An immunofluorescence study: Neurol Res, 2019; 41(3); 234-41
49. Fierabracci A, Pellegrino M, The double role of p53 in cancer and autoimmunity and its potential as therapeutic target: Int J Mol Sci, 2016; 17(12); 1975
50. Erekat NS, Apoptotic mediators are upregulated in the skeletal muscle of chronic/progressive mouse model of Parkinson’s disease: Anat Rec (Hoboken), 2015; 298(8); 1472-78
51. Al-Jarrah MD, Erekat NS, Parkinson disease-induced upregulation of apoptotic mediators could be attenuated in the skeletal muscle following chronic exercise training: NeuroRehabilitation, 2017; 41(4); 823-30
52. Aubrey BJ, Kelly GL, Janic A, How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression?: Cell Death Differ, 2018; 25(1); 104-13
53. Hage-Sleiman R, Bahmad H, Kobeissy H, Genomic alterations during p53-dependent apoptosis induced by gamma-irradiation of Molt-4 leukemia cells: PLoS One, 2017; 12(12); e0190221
54. Yan X, Zhou R, Ma Z, Autophagy-cell survival and death: Adv Exp Med Biol, 2019; 1206; 667-96
55. Li X, He S, Ma B, Autophagy and autophagy-related proteins in cancer: Mol Cancer, 2020; 19(1); 12
56. Guo JY, White E, Autophagy, metabolism, and cancer: Cold Spring Harb Symp Quant Biol, 2016; 81; 73-78
57. Erekat NS, Autophagy precedes apoptosis among at risk cerebellar Purkinje cells in the shaker mutant rat: An ultrastructural study: Ultrastruct Pathol, 2018; 42(2); 162-69
58. Tooze SA, Abada A, Elazar Z, Endocytosis and autophagy: Exploitation or cooperation?: Cold Spring Harb Perspect Biol, 2014; 6(5); a018358
59. Yamahara K, Yasuda M, Kume S, The role of autophagy in the pathogenesis of diabetic nephropathy: J Diabetes Res, 2013; 2013; 193757
60. Kotani T, Kirisako H, Koizumi M, The Atg2-Atg18 complex tethers pre-autophagosomal membranes to the endoplasmic reticulum for autophagosome formation: Proc Natl Acad Sci USA, 2018; 115(41); 10363-68
61. Martin KR, Celano SL, Solitro AR, A potent and selective ULK1 inhibitor suppresses autophagy and sensitizes cancer cells to nutrient stress: iScience, 2018; 8; 74-84
62. Chen Y, He J, Tian M, UNC51-like kinase 1, autophagic regulator and cancer therapeutic target: Cell Prolif, 2014; 47(6); 494-505
63. Wang C, Wang H, Zhang D, Phosphorylation of ULK1 affects autophagosome fusion and links chaperone-mediated autophagy to macroautophagy: Nat Commun, 2018; 9(1); 3492
64. Torii S, Yamaguchi H, Nakanishi A, Identification of a phosphorylation site on Ulk1 required for genotoxic stress-induced alternative autophagy: Nat Commun, 2020; 11(1); 1754
65. You SY, Park YS, Jeon HJ, Beclin-1 knockdown shows abscission failure but not autophagy defect during oocyte meiotic maturation: Cell Cycle, 2016; 15(12); 1611-19
66. Zhang D, Wang W, Sun X, AMPK regulates autophagy by phosphorylating BECN1 at threonine 388: Autophagy, 2016; 12(9); 1447-59
67. Iershov A, Nemazanyy I, Alkhoury C, The class 3 PI3K coordinates autophagy and mitochondrial lipid catabolism by controlling nuclear receptor PPARα: Nat Commun, 2019; 10(1); 1566
68. Xie X, Chen C, Xu CB, The class III PI3K/Beclin-1 autophagic pathway participates in the mmLDL-induced upregulation of ETA receptor in mouse mesenteric arteries: Adv Pharmacol Pharm Sci, 2020; 2020; 5070436
69. Tan Q, Liu Y, Deng X, Autophagy: A promising process for the treatment of acetaminophen-induced liver injury: Arch Toxicol, 2020; 94(9); 2925-38
70. Lystad AH, Simonsen A, Mechanisms and pathophysiological roles of the ATG8 conjugation machinery: Cells, 2019; 8(9); 973
71. Wei Y, Liu M, Li X, Origin of the autophagosome membrane in mammals: Biomed Res Int, 2018; 2018; 1012789
72. Zientara-Rytter K, Subramani S, Mechanistic insights into the role of Atg11 in selective autophagy: J Mol Biol, 2020; 432(1); 104-22
73. Liu H, Dai C, Fan Y, From autophagy to mitophagy: The roles of P62 in neurodegenerative diseases: J Bioenerg Biomembr, 2017; 49(5); 413-22
74. Shin WH, Park JH, Chung KC, The central regulator p62 between ubiquitin proteasome system and autophagy and its role in the mitophagy and Parkinson’s disease: BMB Rep, 2020; 53(1); 56-63
75. Gubbiotti MA, Seifert E, Rodeck U, Metabolic reprogramming of murine cardiomyocytes during autophagy requires the extracellular nutrient sensor decorin: J Biol Chem, 2018; 293(43); 16940-50
76. Cordani M, Donadelli M, Strippoli R, Interplay between ROS and autophagy in cancer and aging: From molecular mechanisms to novel therapeutic approaches: Oxid Med Cell Longev, 2019; 2019; 8794612
77. Wang J, Davis S, Zhu M, Autophagosome formation: Where the secretory and autophagy pathways meet: Autophagy, 2017; 13(5); 973-74
78. Gonzalez A, Hall MN, Lin SC, Hardie DG, AMPK and TOR: The Yin and Yang of cellular nutrient sensing and growth control: Cell Metab, 2020; 31(3); 472-92
79. Garcia D, Shaw RJ, AMPK: Mechanisms of cellular energy sensing and restoration of metabolic balance: Mol Cell, 2017; 66(6); 789-800
80. Herzig S, Shaw RJ, AMPK: Guardian of metabolism and mitochondrial homeostasis: Nat Rev Mol Cell Biol, 2018; 19(2); 121-35
81. Lin S-C, Hardie DG, AMPK: Sensing glucose as well as cellular energy status: Cell Metabolism, 2018; 27(2); 299-313
82. Chauhan AS, Liu X, Jing J, STIM2 interacts with AMPK and regulates calcium-induced AMPK activation: FASEB J, 2019; 33(2); 2957-70
83. Rabanal-Ruiz Y, Otten EG, Korolchuk VI, mTORC1 as the main gateway to autophagy: Essays Biochem, 2017; 61(6); 565-84
84. Li Y, Chen Y, AMPK and autophagy: Adv Exp Med Biol, 2019; 1206; 85-108
85. Jhanwar-Uniyal M, Wainwright JV, Mohan AL, Diverse signaling mechanisms of mTOR complexes: mTORC1 and mTORC2 in forming a formidable relationship: Adv Biol Regul, 2019; 72; 51-62
86. Abraham RT, Making sense of amino acid sensing: Science, 2015; 347(6218); 128-29
87. Angarola B, Ferguson SM, Coordination of Rheb lysosomal membrane interactions with mTORC1 activation: F1000Res, 2020; 9; F1000 F aculty Rev-450
88. Li H, Yu L, Zhao C, Dioscin attenuates highfat dietinduced insulin resistance of adipose tissue through the IRS1/PI3K/Akt signaling pathway: Mol Med Rep, 2019; 19(2); 1230-37
89. Gen S, Matsumoto Y, Kobayashi K-I, Stability of tuberous sclerosis complex 2 is controlled by methylation at R1457 and R1459: Sci Rep, 2020; 10(1); 21160
90. Ribeiro M, López de Figueroa P, Blanco FJ, Insulin decreases autophagy and leads to cartilage degradation: Osteoarthritis Cartilage, 2016; 24(4); 731-39
91. Takahara T, Amemiya Y, Sugiyama R, Amino acid-dependent control of mTORC1 signaling: A variety of regulatory modes: J Biomed Sci, 2020; 27(1); 87
92. Li D, Ding Z, Du K, Reactive oxygen species as a link between antioxidant pathways and autophagy: Oxid Med Cell Longev, 2021; 2021; 5583215
93. Ligeon L-A, Pena-Francesch M, Vanoaica LD, Oxidation inhibits autophagy protein deconjugation from phagosomes to sustain MHC class II restricted antigen presentation: Nat Commun, 2021; 12(1); 1508
94. Liu Y, Wang Z, Xie W, Oxidative stress regulates mitogenactivated protein kinases and cJun activation involved in heat stress and lipopolysaccharideinduced intestinal epithelial cell apoptosis: Mol Med Rep, 2017; 16(3); 2579-87
95. Wang Y, Liu N, Lu B, Mechanisms and roles of mitophagy in neurodegenerative diseases: CNS Neurosci Ther, 2019; 25(7); 859-75
96. Lu N, Li X, Tan R, HIF-1alpha/Beclin1-mediated autophagy is involved in neuroprotection induced by hypoxic preconditioning: J Mol Neurosci, 2018; 66(2); 238-50
97. Dhuriya YK, Sharma D, Necroptosis: A regulated inflammatory mode of cell death: J Neuroinflammation, 2018; 15(1); 199
98. Yu Z, Jiang N, Su W, Zhuo Y, Necroptosis: A novel pathway in neuroinflammation: Front Pharmacol, 2021; 12; 701564
99. Roberts JZ, Crawford N, Longley DB, The role of ubiquitination in apoptosis and necroptosis: Cell Death Differ, 2022; 29(2); 272-84
100. Alicic RZ, Rooney MT, Tuttle KR, Diabetic kidney disease: Challenges, progress, and possibilities: Clin J Am Soc Nephrol, 2017; 12(12); 2032-45
101. Opazo-Rios L, Mas S, Marin-Royo G, Lipotoxicity and diabetic nephropathy: Novel mechanistic insights and therapeutic opportunities: Int J Mol Sci, 2020; 21(7); 2632
102. Wada J, Makino H, Innate immunity in diabetes and diabetic nephropathy: Nat Rev Nephrol, 2016; 12(1); 13-26
103. Yang YY, Deng RR, Chen Z, Piperazine ferulate attenuates high glucoseinduced mesangial cell injury via the regulation of p66(Shc): Mol Med Rep, 2021; 23(5); 374
104. Chen J, Chen JK, Harris RC, EGF receptor deletion in podocytes attenuates diabetic nephropathy: J Am Soc Nephrol, 2015; 26(5); 1115-25
105. Liu M, Liang K, Zhen J, Sirt6 deficiency exacerbates podocyte injury and proteinuria through targeting Notch signaling: Nat Commun, 2017; 8(1); 413
106. Bose M, Almas S, Prabhakar S, Wnt signaling and podocyte dysfunction in diabetic nephropathy: J Investig Med, 2017; 65(8); 1093-101
107. Wang Y, Niu A, Pan Y, Profile of podocyte translatome during development of type 2 and type 1 diabetic nephropathy using podocyte-specific TRAP mRNA RNA-seq: Diabetes, 2021; 70(10); 2377-90
108. Eisenreich A, Leppert U, Update on the protective renal effects of metformin in diabetic nephropathy: Curr Med Chem, 2017; 24(31); 3397-412
109. Bhatti AB, Usman M, Drug targets for oxidative podocyte injury in diabetic nephropathy: Cureus, 2015; 7(12); e393
110. Liu L, Huang S, Xu M, Isoquercitrin protects HUVECs against high glucoseinduced apoptosis through regulating p53 proteasomal degradation: Int J Mol Med, 2021; 48(1); 122
111. Erekat N, Rababa’h R, Al-Jarrah M, Overexpression of renal proapoptotic factors is attenuated subsequent to endurance exercise in Type I diabetes: An immunohistochemistry study: Journal of Natural Science, Biology and Medicine, 2019; 10(1); 24-28
112. Peixoto EB, Papadimitriou A, Teixeira DA, Reduced LRP6 expression and increase in the interaction of GSK3beta with p53 contribute to podocyte apoptosis in diabetes mellitus and are prevented by green tea: J Nutr Biochem, 2015; 26(4); 416-30
113. Zhu X, Xiong X, Yuan S, Validation of the interstitial fibrosis and tubular atrophy on the new pathological classification in patients with diabetic nephropathy: A single-center study in China: J Diabetes Complications, 2016; 30(3); 537-41
114. Schelling JR, Tubular atrophy in the pathogenesis of chronic kidney disease progression: Pediatr Nephrol, 2016; 31(5); 693-706
115. Satou R, Cypress MW, Woods TC, Blockade of sodium-glucose cotransporter 2 suppresses high glucose-induced angiotensinogen augmentation in renal proximal tubular cells: Am J Physiol Renal Physiol, 2020; 318(1); F67-F75
116. Ding M, Tang Z, Liu W, Burdock fructooligosaccharide attenuates high glucose-induced apoptosis and oxidative stress injury in renal tubular epithelial cells: Front Pharmacol, 2021; 12; 784187
117. Liao J, Liu B, Chen K, Galangin attenuates oxidative stress-mediated apoptosis in high glucose-induced renal tubular epithelial cells through modulating renin-angiotensin system and PI3K/AKT/mTOR pathway: Toxicol Res (Camb), 2021; 10(3); 551-60
118. Sun Y, Ge X, Li X, High-fat diet promotes renal injury by inducing oxidative stress and mitochondrial dysfunction: Cell Death Dis, 2020; 11(10); 914
119. Priante G, Gianesello L, Ceol M, Cell death in the kidney: Int J Mol Sci, 2019; 20(14); 3598
120. Asadipooya K, Uy EM, Advanced glycation end products (AGEs), receptor for AGEs, diabetes, and bone: Review of the literature: J Endoc Soc, 2019; 3(10); 1799-818
121. Takahashi A, Takabatake Y, Kimura T, Autophagy inhibits the accumulation of advanced glycation end products by promoting lysosomal biogenesis and function in the kidney proximal tubules: Diabetes, 2017; 66(5); 1359-72
122. Audzeyenka I, Bierzynska A, Piwkowska A, Viability of primary cultured podocytes is associated with extracellular high glucose-dependent autophagy downregulation: Mol Cell Biochem, 2017; 430(1-2); 11-19
123. Bork T, Liang W, Yamahara K, Podocytes maintain high basal levels of autophagy independent of mtor signaling: Autophagy, 2020; 16(11); 1932-48
124. Yang D, Livingston MJ, Liu Z, Autophagy in diabetic kidney disease: Regulation, pathological role and therapeutic potential: Cell Mol Life Sci, 2018; 75(4); 669-88
125. Lin TA, Wu VC, Wang CY, Autophagy in chronic kidney diseases: Cells, 2019; 8(1); 61
126. Yin L, Yu L, He JC, Chen A, Controversies in podocyte loss: Death or detachment?: Front Cell Dev Biol, 2021; 9; 771931
127. Koch EAT, Nakhoul R, Nakhoul F, Nakhoul N, Autophagy in diabetic nephropathy: A review: Int Urol Nephrol, 2020; 52(9); 1705-12
128. Liu WJ, Gan Y, Huang WF, Lysosome restoration to activate podocyte autophagy: A new therapeutic strategy for diabetic kidney disease: Cell Death Dis, 2019; 10(11); 806
129. Mizunoe Y, Kobayashi M, Tagawa R, Association between lysosomal dysfunction and obesity-related pathology: A key knowledge to prevent metabolic syndrome: Int J Mol Sci, 2019; 20(15); 3688
130. Yasuda-Yamahara M, Kume S, Emerging role of podocyte autophagy in the progression of diabetic nephropathy: Autophagy, 2015; 11(12); 2385-86
131. Kitada M, Ogura Y, Monno I, Koya D, Regulating autophagy as a therapeutic target for diabetic nephropathy: Curr Diab Rep, 2017; 17(7); 53
132. Kaushal GP, Shah SV, Autophagy in acute kidney injury: Kidney Int, 2016; 89(4); 779-91
133. Ozkok A, Edelstein CL, Pathophysiology of cisplatin-induced acute kidney injury: Biomed Res Int, 2014; 2014; 967826
134. Sugawara H, Moniwa N, Kuno A, Activation of the angiotensin II receptor promotes autophagy in renal proximal tubular cells and affords protection from ischemia/reperfusion injury: J Pharmacol Sci, 2021; 145(2); 187-97
135. Guillén C, Benito M, mTORC1 overactivation as a key aging factor in the progression to type 2 diabetes mellitus: Front Endocrinol (Lausanne), 2018; 9; 621
136. Meng XM, Ren GL, Gao L, NADPH oxidase 4 promotes cisplatin-induced acute kidney injury via ROS-mediated programmed cell death and inflammation: Lab Invest, 2018; 98(1); 63-78
137. Fang Y, Shen X, Ubiquitin carboxyl-terminal hydrolases: Involvement in cancer progression and clinical implications: Cancer Metastasis Rev, 2017; 36(4); 669-82
138. Zhang Y, Xu C, Ye Q, Podocyte apoptosis in diabetic nephropathy by BASP1 activation of the p53 pathway via WT1: Acta Physiol (Oxf), 2021; 232(1); e13634
139. Zhang Y, Chen X, Fan Y, XCL1 aggravates diabetic nephropathy-mediated renal glomerular endothelial cell apoptosis and inflammatory response via regulating p53/nuclear factor-kappa B pathway: Nephron, 2022; 146(1); 84-98
140. Ahmed OM, Ali TM, Abdel Gaid MA, Elberry AA, Effects of enalapril and paricalcitol treatment on diabetic nephropathy and renal expressions of TNF-alpha, p53, caspase-3 and Bcl-2 in STZ-induced diabetic rats: PLoS One, 2019; 14(9); e0214349
141. Tome ME, Lutz NW, Briehl MM, Overexpression of catalase or Bcl-2 alters glucose and energy metabolism concomitant with dexamethasone resistance: Biochim Biophys Acta, 2004; 1693(1); 57-72
142. Abdo S, Shi Y, Otoukesh A, Catalase overexpression prevents nuclear factor erythroid 2-related factor 2 stimulation of renal angiotensinogen gene expression, hypertension, and kidney injury in diabetic mice: Diabetes, 2014; 63(10); 3483-96
143. Shi Y, Lo CS, Chenier I, Overexpression of catalase prevents hypertension and tubulointerstitial fibrosis and normalization of renal angiotensin-converting enzyme-2 expression in Akita mice: Am J Physiol Renal Physiol, 2013; 304(11); F1335-46
144. Verzola D, Bertolotto MB, Villaggio B, Taurine prevents apoptosis induced by high ambient glucose in human tubule renal cells: J Investig Med, 2002; 50(6); 443-51
145. Fan H, Zhang W, Overexpression of Linc 4930556M19Rik suppresses high glucose-triggered podocyte apoptosis, fibrosis and inflammation via the miR-27a-3p/metalloproteinase 3 (TIMP3) axis in diabetic nephropathy: Med Sci Monit, 2020; 26; e925361
146. Robertson LT, Trevino-Villarreal JH, Mejia P, Protein and calorie restriction contribute additively to protection from renal ischemia reperfusion injury partly via leptin reduction in male mice: J Nutr, 2015; 145(8); 1717-27
147. Gouda K, AbdelHamid S, Mansour A, Amelioration of diabetic nephropathy by targeting autophagy via rapamycin or fasting: relation to cell apoptosis/survival: Curr Issues Mol Biol, 2021; 43(3); 1698-714
148. Liu L, Yang L, Chang B, The protective effects of rapamycin on cell autophagy in the renal tissues of rats with diabetic nephropathy via mTOR-S6K1-LC3II signaling pathway: Ren Fail, 2018; 40(1); 492-97
149. Liu WJ, Huang WF, Ye L, The activity and role of autophagy in the pathogenesis of diabetic nephropathy: Eur Rev Med Pharmacol Sci, 2018; 22(10); 3182-89
150. Borges CM, Fujihara CK, Malheiros D, Metformin arrests the progression of established kidney disease in the subtotal nephrectomy model of chronic kidney disease: Am J Physiol Renal Physiol, 2020; 318(5); F1229-F36
151. Ren H, Shao Y, Wu C, Metformin alleviates oxidative stress and enhances autophagy in diabetic kidney disease via AMPK/SIRT1-FoxO1 pathway: Mol Cell Endocrinol, 2020; 500; 110628
152. Park HS, Lim JH, Kim MY, Resveratrol increases AdipoR1 and AdipoR2 expression in type 2 diabetic nephropathy: J Transl Med, 2016; 14(1); 176
153. He T, Xiong J, Nie L, Resveratrol inhibits renal interstitial fibrosis in diabetic nephropathy by regulating AMPK/NOX4/ROS pathway: J Mol Med (Berl), 2016; 94(12); 1359-71
154. Li A, Yi B, Han H, Vitamin D-VDR (vitamin D receptor) regulates defective autophagy in renal tubular epithelial cell in streptozotocin-induced diabetic mice via the AMPK pathway: Autophagy, 2021; 18(4); 877-90
155. Lim JH, Kim HW, Kim MY, Cinacalcet-mediated activation of the CaMKKbeta-LKB1-AMPK pathway attenuates diabetic nephropathy in db/db mice by modulation of apoptosis and autophagy: Cell Death Dis, 2018; 9(3); 270
156. Yi W, OuYang Q, Adiponectin improves diabetic nephropathy by inhibiting necrotic apoptosis: Arch Med Sci, 2019; 15(5); 1321-28
Figures
Figure 1. Extrinsic and intrinsic pathways of apoptosis. The extrinsic apoptotic pathway is triggered by the binding of a particular death ligand to death receptor, leading to its trimerization and the consequent recruitment of procaspase-8, which is subsequently activated. In contrast, the intrinsic apoptotic pathway starts in response to apoptotic stimuli that induce pro-apoptotic proteins, such as B-cell lymphoma 2-associated X protein (Bax), triggering the permeabilization of the outer mitochondrial membrane, and the consequent release of cytochrome c into the cytosol, where it contributes to the formation of a multimeric apoptotic peptidase activating factor 1 (Apaf-1)/cytochrome c complex that recruits procaspase-9, forming the apoptosome. Hence, procaspase-9 is activated and consequently detached from the complex. Ultimately, active caspase-8 and active caspase-9 activate executioner caspases-3, -6, and/or -7, which mediate apoptosis. This figure was adapted with permission from John Wiley and Sons, from “Apoptosis and its therapeutic implications in neurodegenerative diseases” by Nour S. Erekat, 2022, Clinical Anatomy [34].Figure 2. The process of autophagy. The autophagic process occurs via 5 major stages, which are initiation, nucleation, elongation/closure, fusion, and breakdown. Initiation stage: The unc51-like kinase 1 (Ulk1) complex consists of Ulk1 serine/threonine protein kinase, autophagy-related gene 13 (Atg13), and focal adhesion kinase family interacting protein of 200 kD (FIP200). Ulk1 is required for the initiation stage. 2. Nucleation stage: Beclin 1, which exists with class III phosphoinositide 3-kinase (PI3K), is essential for the nucleation stage. 3. Elongation/closure stage: A membrane sac isolation membrane wraps around the degradation substrates and ultimately closes to become an autophagosome. This stage requires the microtubule-associated protein light chain 3 (LC3). 4. Fusion stage: the outer membrane of the autophagosome completely fuses with the outer lysosomal membrane. 5. Breakdown stage: contents of the autophagosome are released and subsequently degraded into the lysosomal cavity. This figure was generated using Microsoft PowerPoint Software, version 10, Microsoft Corp., USA.Figure 3. The role of hyperglycemia in the suppression of autophagy and induction of apoptosis and necroptosis in diabetic nephropathy. Hyperglycemia causes overproduction of advanced glycation end-products (AGEs), which inhibit autophagy, leading to diabetic nephropathy. Hyperglycemia induces reactive oxygen species generation, which initiates apoptosis. Hyperglycemia also increases tumor suppressor p53 messenger ribonucleic acid (mRNA) and protein expression, thus activating the transcription of many pro-apoptotic proteins that stimulate the extrinsic and the intrinsic apoptotic pathways. Additionally, hyperglycemia stimulates renin-angiotensin, which is involved in apoptosis and is involved in diabetic nephropathy. Hyperglycemia can also trigger necroptosis in diabetic nephropathy via activating the interacting serine/threonine kinase 1 (RIPK1)- RIPK3 signaling pathway. This figure was generated using Microsoft PowerPoint Software, version 10, Microsoft Corp., USA. In Press
07 Mar 2024 : Clinical Research
Knowledge of and Attitudes Toward Clinical Trials: A Questionnaire-Based Study of 179 Male Third- and Fourt...Med Sci Monit In Press; DOI: 10.12659/MSM.943468
08 Mar 2024 : Animal Research
Modification of Experimental Model of Necrotizing Enterocolitis (NEC) in Rat Pups by Single Exposure to Hyp...Med Sci Monit In Press; DOI: 10.12659/MSM.943443
18 Apr 2024 : Clinical Research
Comparative Analysis of Open and Closed Sphincterotomy for the Treatment of Chronic Anal Fissure: Safety an...Med Sci Monit In Press; DOI: 10.12659/MSM.944127
08 Mar 2024 : Laboratory Research
Evaluation of Retentive Strength of 50 Endodontically-Treated Single-Rooted Mandibular Second Premolars Res...Med Sci Monit In Press; DOI: 10.12659/MSM.944110
Most Viewed Current Articles
17 Jan 2024 : Review article
Vaccination Guidelines for Pregnant Women: Addressing COVID-19 and the Omicron VariantDOI :10.12659/MSM.942799
Med Sci Monit 2024; 30:e942799
14 Dec 2022 : Clinical Research
Prevalence and Variability of Allergen-Specific Immunoglobulin E in Patients with Elevated Tryptase LevelsDOI :10.12659/MSM.937990
Med Sci Monit 2022; 28:e937990
16 May 2023 : Clinical Research
Electrophysiological Testing for an Auditory Processing Disorder and Reading Performance in 54 School Stude...DOI :10.12659/MSM.940387
Med Sci Monit 2023; 29:e940387
01 Jan 2022 : Editorial
Editorial: Current Status of Oral Antiviral Drug Treatments for SARS-CoV-2 Infection in Non-Hospitalized Pa...DOI :10.12659/MSM.935952
Med Sci Monit 2022; 28:e935952