26 July 2020: Meta-Analysis
Integrated Analysis of Three Publicly Available Gene Expression Profiles Identified Genes and Pathways Associated with Clear Cell Renal Cell Carcinoma
YuPing Han1ACD, LinLin Wang2EF, Ye Wang3AB*DOI: 10.12659/MSM.919965
Med Sci Monit 2020; 26:e919965






Abstract
BACKGROUND: Although advances have been achieved in the therapy of clear cell renal cell carcinoma (ccRCC), the pathogenesis of ccRCC is not yet fully understood. This study aimed to explore the critical genes and pathways associated with ccRCC by meta-analysis.
MATERIAL AND METHODS: We performed an integrated analysis of 3 publicly available microarray datasets developed from ccRCC tumor samples and normal tissues. A list of overlapped differentially expressed genes (DEGs) with the consistent expression trend in ccRCC tumor samples were identified, for which the protein–protein interaction (PPI) network was constructed, followed by topology structure and module analysis. The microRNA (miRNA) regulatory network and ccRCC associated pathway network were reconstructed.
RESULTS: A total of 504 genes were found to be consistently and differentially regulated based on 3 microarray datasets. The overrepresented pathways for DEGs included citric acid cycle (TCA cycle) and peroxisome proliferator-activated receptor (PPAR) signaling pathway and cell cycle. The PPI network was clustered into 6 modules that were closely related with the M phase, desmosome assembly, and response to hormone stimulus. The hsa04110: cell cycle and hsa04510: focal adhesion were the significant pathways associated with ccRCC overlapped with enrichment analysis. KDR and ITGB4 were focal-adhesion-associated genes, which were regulated by has-miR-424 and has-miR-204, respectively. CCND2 and CCNA2 were cell-cycle-associated genes, which were regulated by hsa-miR-324-3p, hsa-miR-146a and hsa-miR-145.
CONCLUSIONS: Cell cycle and focal adhesion were dysregulated in ccRCC, which were associated with the expression of CCND2, ITGB4, KDR, and CCNA2 genes. The deregulation of pathways and associated genes may provide insights to ccRCC research and therapy.
Keywords: clear cell renal cell carcinoma, Focal Adhesion, microRNA, Pathway-Associated Genes, Biomarkers, Tumor, Carcinoma, Renal Cell, Databases, Genetic, Gene Regulatory Networks, Kidney Neoplasms, Protein Interaction Maps
Background
Renal cell carcinoma (RCC) is a type of solid tumor derived from the renal epithelium, which accounts for 80–85% of all renal cancers [1]. Clear cell renal cell carcinoma (ccRCC) is the most common type of RCC and characterized by complex histological changes and metastatic potential. Among the various subtypes of RCC, ccRCC is closely associated with poor outcomes and cancer-related deaths [2]. Most ccRCCs are diagnosed sporadically. Although marked advancement has been achieved in RCC treatment, ccRCCs are refractory to conventional chemotherapy. It has been proposed that a good understanding of the preoperative characteristics of renal cancers may improve the therapeutic management and prognosis [3,4].
Although the pathogenesis of ccRCC has not been fully elucidated, oncogenic metabolism and epigenetic reprogramming are the central features of ccRCC progression and development. DNA microarray technology and high-throughput sequencing have been widely used in cancer profiling and identifying biomarkers for cancers [2,5,6]. Sato et al. performed an integrated molecular study of ccRCC by whole-genome/exome and RNA sequencing and found that p53-related pathways and mRNA processing were significant in ccRCC [7]. In another integrated molecular study, the PI(3)K/AKT pathway was proposed to be the target for ccRCC treatment [8]. It has been reported that ccRCC is characterized by the loss expression of Von Hippel-Lindau (VHL) tumor suppressor gene which is implicated in angiogenesis, apoptosis, and glycolysis [9,10]. Hakimi et al. performed metabolomic profiling combined with transcriptomic expression profiling of ccRCC, suggesting that the dysregulation of oxidative phosphorylation and amino acid metabolism was involved in ccRCC development [11].
Currently, the management for ccRCC is recommended based on the histology of tumor cells [12]. The cytoreductive nephrectomy is recommended to patients in early stage disease, and treatment with bevacizumab (combined with interferon), sunitinib and pazopanib has been proposed to have efficacy as first line treatment for ccRCC patients. A number of studies have revealed that ccRCC is characterized by metabolic reprogramming. Drugs targeting metabolic reprogramming have been suggested to be novel treatment for ccRCC and their efficacy has been evaluated under clinical trials.
Expression profiling studies can identify the target genes or pathways for disease treatment. A recent study of bioinformatics analysis revealed that chemokine signaling, and the complement and coagulation cascade were key pathways in ccRCC [13]. Yang et al. performed multi-tool joint analysis and suggested that TF and B4GALNT1 were associated with ccRCC metastasis and were prognostic biomarkers [14]. It is necessary to validate the specific genes or pathways screened based on the microarray data or sequencing profiles by experiments.
In our study, we performed an integrated analysis of microarray datasets related with ccRCC from 3 independent studies. All 3 studies compared the gene expression profiles of ccRCC tumor samples with normal tissues. We performed interstudy validation of differentially expressed genes (DEGs) from 3 independent datasets and reconstructed the gene and pathway network. We expected that inter-validated sets of dysregulated genes and pathways could provide clues for understanding ccRCC.
Material and Methods
MICROARRAY DATASET COLLECTION:
Three microarray datasets related to ccRCC were retrieved from the publicly available Gene Expression Omnibus (GEO) [17] database (http://www.ncbi.nlm.nih.gov/geo/) at the National Center for Biotechnology Information (NCBI), including GSE6344 [18,19], GSE781 [20], and GSE53000 [21]. The GSE6344 dataset included 10 ccRCC tumor tissues and 10 paired normal tissues. The GSE781 dataset contained 9 ccRCC tumor samples and 8 normal tissue samples. These samples were measured based on the platform of GPL96 [HG-U133A] Affymetrix Human Genome U133A Array. The GSE53000 dataset, consisting of 56 ccRCC tumor samples and 6 normal samples, were produced based on the platform of [HuGene-1_0-st] Affymetrix Human Gene 1.0 ST Array.
DATA PREPROCESSING:
The raw data of 3 Affymetrix microarrays were downloaded and preprocessed using R 3.4.1 oligo package version 3.6 (http://www.bioconductor.org/packages/release/bioc/html/oligo.html), which included background correction and gene expression pattern normalization [22].
GENES WITH DIFFERENTIAL EXPRESSION:
The DEGs in multiple datasets were analyzed using MetaDE.ES function in MetaDE package version 1.0.5 (23) in R 3.4.1 (
Then, the genes of interest were subjected to Gene Ontology (GO) function and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis based on the Fisher’s exact test by the database for Annotation, Visualization and Integrated Discovery (DAVID) [24] online tool (https://david.ncifcrf.gov/). FDR <0.05 was set as the cutoff value.
PROTEIN–PROTEIN INTERACTION (PPI) NETWORK:
The interactions of proteins encoded by DEGs were retrieved using the Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) [25] version 10.5 (https://string-db.org/). The protein pairs with interaction score >0.8 were collected. The PPI network was visualized using Cytoscape [26] 3.6.1 (http://www.cytoscape.org/).
PPI NETWORK STRUCTURE ANALYSIS:
For the scale-free properties of the PPI network, the topology of the network was analyzed, including the node degree, betweenness centrality (BC), and closeness centrality (CC). The node degree indicated the number of links of a highly connected node to other nodes. BC of nodes is widely analyzed in a large complex network based on shortest paths [27]. BC was calculated using the following formula:
σst indicates the shortest path between s and t, and σst (v) is the number of links to node v. BC ranges from 0 to 1. The closeness to 1 indicates the high centrality measure.
CC is defined as the closeness of a given node from all other nodes [27] and calculated as follows:
V indicates the node set, t is a node in the node set, and dG(V,T) is the sum of the distance of paths. CC ranges from 0 to 1, and the closeness to 1 indicates the high centrality measure.
MODULE ANALYSIS:
Genes that are clustered to one module may have a similar biological function. The modules of PPI network were measured using Cytoscape plugin Molecular Complex Detection (Mcode). The cutoff degree was set to 2, cutoff node score was set to 0.2 and K-core was set to 2. The functions of modules were annotated using Biological Network Gene Ontology plugin with adj
PREDICTION OF CCRCC-RELATED MIRNA:
The ccRCC-related microRNA (miRNA) were retrieved from the Renal Cancer Gene Database (RCDB) [28] (http://www.juit.ac.in/attachments/jsr/rcdb/homenew.html). The experiment validated targets of ccRCC-related miRNAs were downloaded from miRWalk2 database [29] (http://zmf.umm.uni-heidelberg.de/apps/zmf/mirwalk2/). The DEGs that overlapped with miRNA targets were selected for ccRCC miRNA-DEG target regulatory network construction. The genes of interest in the regulatory network were subjected to GO function and KEGG pathway analysis using the DAVID online tool.
CCRCC-RELATED PATHWAY NETWORK CONSTRUCTION:
The KEGG pathways related with ccRCC were retrieved from the Comparative Toxicogenomics Database 2017 update [30] (http://ctd.mdibl.org/) with the keywords of clear cell renal cell carcinoma. The pathways that significantly enriched by miRNA targets were highlighted for ccRCC-related pathway network construction.
Results
IDENTIFICATION OF DEGS IN CCRCC TUMOR TISSUES COMPARED WITH NORMAL ONES:
After the expression data were normalized, a total of 504 DEGs (169 downregulated genes and 335 upregulated genes) were identified by MetaDE package based on three datasets.
To understand the molecular functions of DEGs, GO function and KEGG pathway analysis were performed. The downregulated genes were significantly enriched in 31 GO and pathway terms including 8 GO-Biological Process (BP) terms, 12 GO-Cellular Component (CC) terms, 4 GO-Molecular Function (MF) terms, and 7 KEGG pathways. The upregulated genes were closely related to 11 GO-CC functions, 8 GO-MFs, and 7 pathways. The detailed information is shown in Figure 1. The overrepresented pathways of downregulated genes mainly included hsa00020: citrate cycle (TCA cycle), hsa03320: peroxisome proliferator-activated receptor (PPAR) signaling pathway, and hsa00071: fatty acid metabolism. The dysregulated pathways involving upregulated genes mainly included hsa04610: complement and coagulation cascades, hsa04666: Fc gamma R-mediated phagocytosis, and hsa04110: cell cycle.
PPI NETWORK:
As shown in Figure 2, the PPI network comprising of 621 edges connecting 257 gene nodes was constructed. There were 59 downregulated genes and 198 upregulated genes in the PPI network.
PPI NETWORK TOPOLOGY ANALYSIS:
Based on 3 topological parameters, the hub genes with high centrality in the PPI network were mined. As shown in Table 1, the top 10 hub genes are presented, including CDK1, QSOX1, CCNA2, and AURKB.
MODULE ANALYSIS:
With the application of Mcode, 6 function modules were obtained in the PPI network (Figure 3). The detailed information of modules is shown in Table 2. Module 1 was the most significant cluster (score=6) with 13 nodes and 78 edges. Function annotation indicated that module 1–6 were closely related to 13, 9, 13, 10, 12, and 12 GO-BP terms, respectively, including M phase, coagulation, signaling, desmosome assembly, protein modification by small protein conjugation, and response to hormone stimulus.
CCRCC-ASSOCIATED MIRNA REGULATORY NETWORK:
Based on the information of RCDB, there were 60 records of ccRCC-related miRNAs. Combined with the miRNA targets recorded in the miRWalk2 database, the miRNA regulatory network with 20 miRNA-DEG target interactions was constructed (Figure 4). After function and pathway analysis, the gene targets in the miRNA regulatory network were mainly enriched in 16 GO-BP terms and 2 pathways, including cell cycle phase (GO: 0022403), hemopoiesis (GO: 0030097), immune system development (GO: 0002520), focal adhesion (hsa04510), and cell cycle (hsa04110) pathway (Table 3).
CCRCC-ASSOCIATED PATHWAY NETWORK:
There were 191 ccRCC-related pathways in the CTD database, among which focal adhesion (hsa04510) and cell cycle (hsa04110) were overrepresented by miRNA-target genes including CCND2, ITGB4, KDR, and CCNA2. As shown in Figure 5, the ccRCC-associated pathway network was visualized. Has-miR-424 targeting KDR, has-miR-204 targeting ITGB4, and has-miR-324-3p targeting CCND2 were involved in the focal adhesion pathway. CCND2 regulated by hsa-miR-324-3p and CCNA2 regulated by has-miR-146a and hsa-miR-145 were enriched in the cell cycle pathway.
Discussion
ccRCC is the most common subtype of RCCs, which has been highlighted by the poor prognosis and metastatic potential. The molecular genetic profile of ccRCC has not been clarified. The increasing availability and development of DNA microarray technology has facilitated cancer profiling studies. In this study, we performed an integrated analysis of 3 independent microarray datasets related to ccRCC and provided the targets for future research and therapy for ccRCC.
With the application of an interstudy cross-validation approach, a cohort of 504 genes was identified to be consistently dysregulated in ccRCC based on 3 independent microarray datasets. The pathway analysis showed that hsa00020: citrate cycle (TCA cycle), hsa03320: PPAR signaling pathway, and hsa04110: cell cycle were the significant pathways dysregulated by DEGs in ccRCC. Similar findings were found in the differentially expressed proteins in RCC tissues compared with normal tissues based on proteomics-based approaches [31]. In a previous study, TCA cycle and PPAR signaling pathways were found to be the important enriched pathways in 596 differentially expressed proteins in RCC using 3 available pathway analysis tools. Evidence from a recent study also showed that the PPARα gene was a diagnostic and prognostic biomarker for ccRCC [32], which supports the significant role of PPAR signaling pathway in ccRCC. All these aforementioned findings confirmed that our findings were significant. In addition, a previous study that mined published cancer-related microarray datasets identified that the differentially regulated genes played a critical role in cell cycle control [33], as measured by the pathway analysis of DEGs in this study. Moreover, hsa04110: cell cycle and hsa04510: focal adhesions were found to be the ccRCC-related pathways that overlapped with the enrichment pathways of GO categories. It has been reported that the cell cycle regulator B-cell translocation gene 2 (BTG2) was dysregulated in ccRCC, which played a key role in RCC development [34]. A pathway-based candidate gene evaluation study suggested that the cell cycle was the most significant pathway implicated with CCND 2 gene associated with lung cancer [35]. Our data showed that CCND2 and CCNA2 were the cell-cycle-associated genes in ccRCC, which were upregulated in tumor samples compared with normal tissues. The D-type cyclins were cell-cycle-related proteins, which were involved in G1/S phase transition [36]. CCND2 is a D-type cyclin gene, which is found to be upregulated in various cancers and implicated in cell proliferation and cell cycle control [37]. The overexpression of CCND2 has been shown to promote cell proliferation and cell cycle progression in non-small cell lung cancer (NSCLC) cells [38]. In a recent study, Luo et al. identified CCND1 to be a potential prognostic biomarker of ccRCC by bioinformatic analysis [39]. Similar to CCND2, CCND1 is another member of D-type cyclin genes which is a protooncogene involved in cell cycle regulation. Thus, we speculated that CCND2 plays a key role in cancer development.
The increased levels of cell cycle associated genes are stabilized due to downregulation of specific miRNAs. MiR-146a-5p was found to inhibit cell cycle in a NSCLC cell line by targeting CCND2 expression [38]. MiR-154 inhibits cell proliferation in prostate cancer by suppressing CCND2 expression [40]. Our data showed that hsa-miR-324-3p plays a regulatory role in the cell cycle of ccRCC by targeting CCND2. It is reported that hsa-miR-324-3p is a specific miRNA in ccRCC relative to papillary RCC by miRNA profiling analysis [41]. Generally, hsa-miR-324-3p may play a tumor suppressor role in ccRCC by targeting CCND2.
Furthermore, CCNA2 was found to be the hub gene with high centrality in the PPI network. CCNA2, regulated by hsa-miR-146a and hsa-miR-145, was significantly associated with the cell cycle pathway. The cyclins of the CCNA family genes were implicated in G2-M transition and CCNA2 played a regulatory role in proteolytic control of cell cycle progression during M phrase [42]. A TCGA and GEO-based study suggested that the downregulation of hsa-miR-146a had tumor suppressive effects on hepatocellular carcinoma [43]. CCNA2 has been identified to be the target for miR-145-5p in prostate cancer cells by bioinformatic and function analysis [44]. The overexpression of miR-145-5p has been reported to inhibit prostate cancer cell proliferation [45]. Thus, hsa-miR-146a and hsa-miR-145 may inhibit the cell cycle pathway in ccRCC by targeting CCNA2.
The focal adhesion pathway plays a key role in cell proliferation, survival, and migration and has been suggested as the therapeutic target for cancer [46,47]. Kinase insert domain receptor (KDR) is required for vinculin assembly in focal adhesion plaque [48]. ITGB4 (integrin β4), as a member of integrin genes, is involved in tumor cell migration and has been supported to be the prognostic marker for colon cancer [49]. The increased expression of ITGB4 is related to cell growth, survival, and proliferation and predicts the development of renal cancer [50]. In this study, hsa-miR-424 and hsa-miR-204 are found to be associated with focal adhesion by targeting KDR and ITGB4, respectively. Regulation of hsa-miR-424 and hsa-miR-204 expression may control ccRCC development by mediating the focal adhesion pathway.
Although our findings may provide new perspective in understanding the pathogenesis of ccRCC and discovery of novel therapy, there were some limitations in our study. Firstly, only 3 microarray datasets were included in our study and the dataset size was relatively small. Besides, for the limitation of materials, we cannot provide further functional validation of the critical genes identified in our study. Thus, lacking experimental validation was a limitation in this study.
Conclusions
Cell cycle and focal adhesion were found to be the significant pathways in ccRCC, which were generated by overlapping the information in CTD and pathway enrichment. CCNA2 and CCND2 were the cell-cycle-associated genes, and KDR and ITGB4 were the focal-adhesion-associated genes. Regulation of the expression of miRNAs may provide insights to ccRCC research and therapy in the near future.
Figures
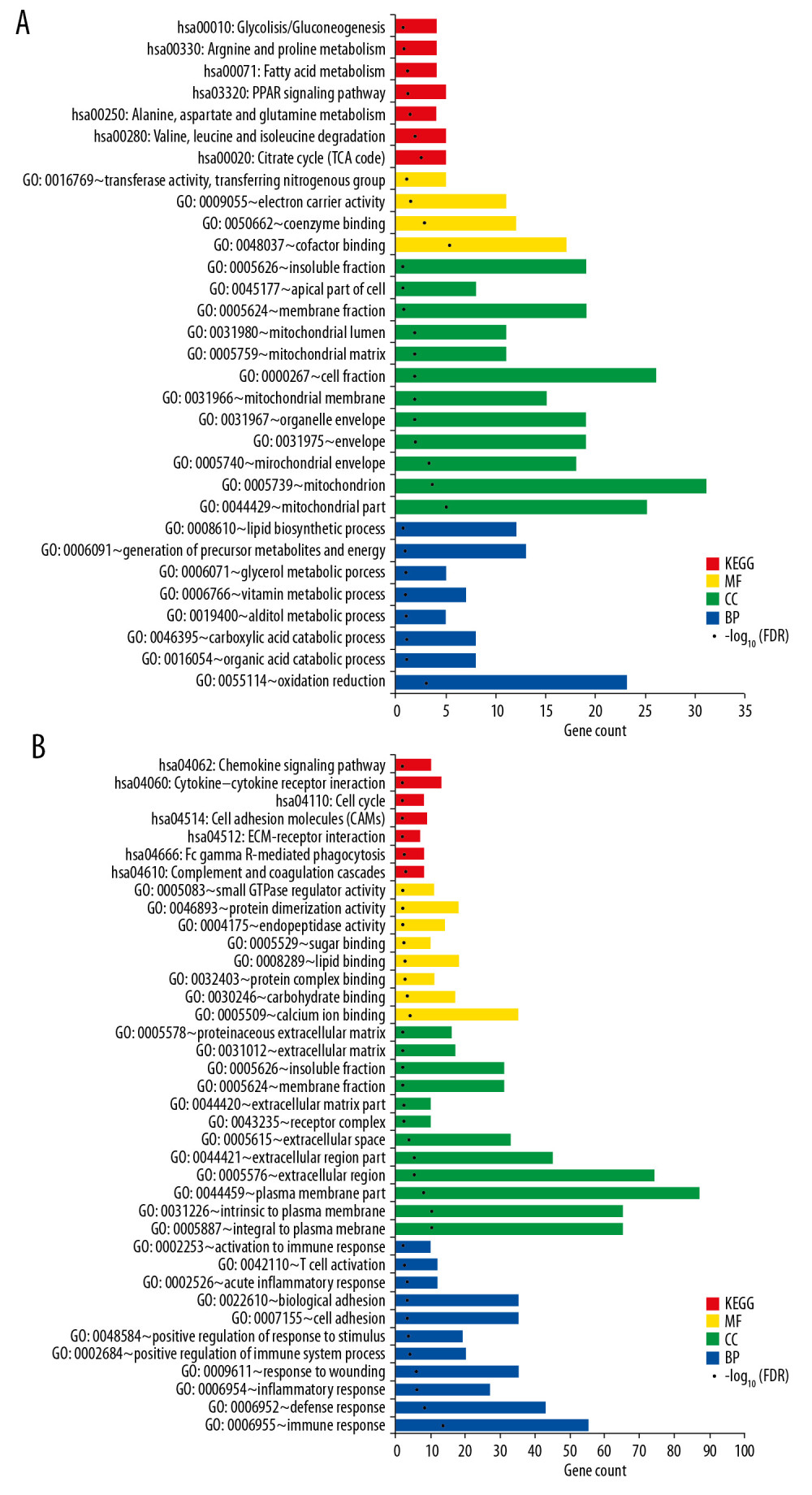 Figure 1. The significant GO function and pathways for downregualted genes (A) and upregulated genes (B). The downregulated and upregulated genes were subjected to GO function and pathway enrichment analysis by Fisher’s exact test. Red – KEGG pathway; yellow – molecular function; green – cell component; blue – biological process. GO – gene ontology; KEGG – Kyoto Encyclopedia of Genes and Genomes.
Figure 1. The significant GO function and pathways for downregualted genes (A) and upregulated genes (B). The downregulated and upregulated genes were subjected to GO function and pathway enrichment analysis by Fisher’s exact test. Red – KEGG pathway; yellow – molecular function; green – cell component; blue – biological process. GO – gene ontology; KEGG – Kyoto Encyclopedia of Genes and Genomes. 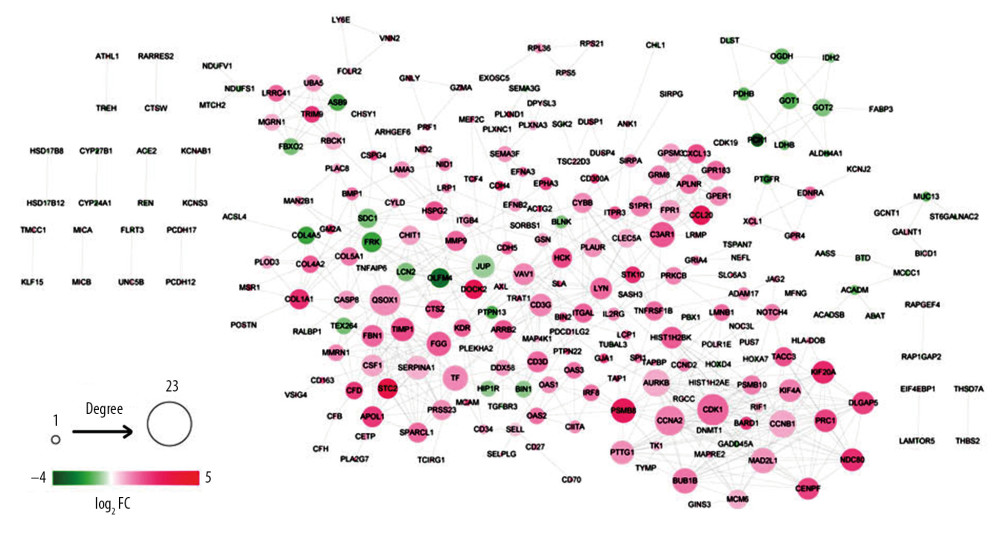 Figure 2. PPI network. The differentially expressed genes were mapped to proteins based on STRING database. PPI network was visualized using Cytoscape software. Green – downregulated genes; red – upregulated genes. The size of node indicates the node degree. PPI – protein–protein interaction; STRING – Search Tool for the Retrieval of Interacting Genes/Proteins.
Figure 2. PPI network. The differentially expressed genes were mapped to proteins based on STRING database. PPI network was visualized using Cytoscape software. Green – downregulated genes; red – upregulated genes. The size of node indicates the node degree. PPI – protein–protein interaction; STRING – Search Tool for the Retrieval of Interacting Genes/Proteins. 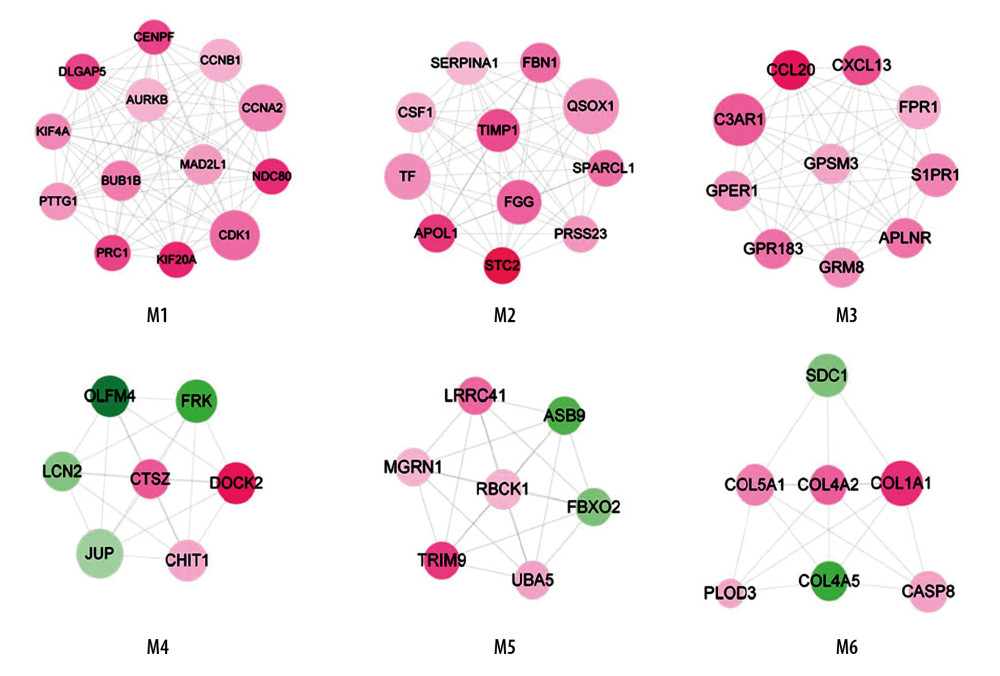 Figure 3. Modules in PPI network. The modules in PPI network were analyzed using Cytoscape plugin Molecular Complex Detection (Mcode). With cutoff degree ≥2, cutoff node score ≥0.2 and K-core ≥2, 6 modules were obtained. Green – downregulated genes; red – upregulated genes. The size of node indicates the node degree. PPI – protein–protein interaction.
Figure 3. Modules in PPI network. The modules in PPI network were analyzed using Cytoscape plugin Molecular Complex Detection (Mcode). With cutoff degree ≥2, cutoff node score ≥0.2 and K-core ≥2, 6 modules were obtained. Green – downregulated genes; red – upregulated genes. The size of node indicates the node degree. PPI – protein–protein interaction. 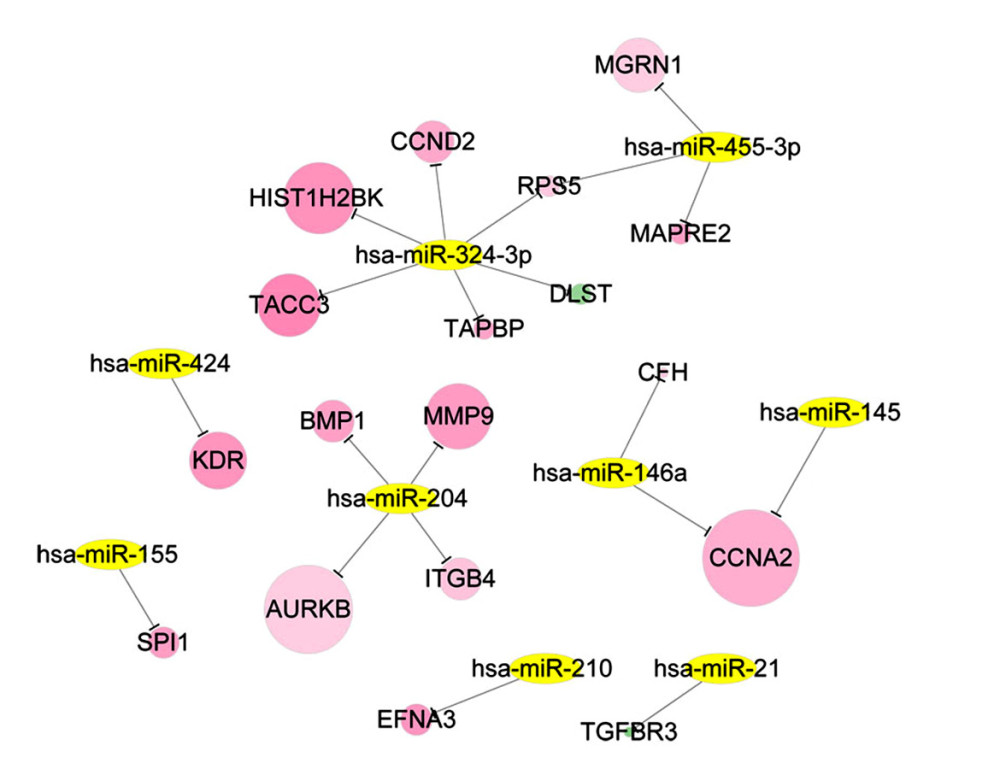 Figure 4. The ccRCC associated miRNA regulatory network. The ccRCC-related miRNAs were retrieved from the Renal Cancer Gene Database and DEGs that overlapped with miRNA targets were selected for ccRCC miRNA-DEG target regulatory network construction. Green – downregulated genes; red – upregulated genes; yellow – miRNAs. ccRCC – clear cell renal cell carcinoma; miRNA – microRNA; DEGs – differentially expressed genes.
Figure 4. The ccRCC associated miRNA regulatory network. The ccRCC-related miRNAs were retrieved from the Renal Cancer Gene Database and DEGs that overlapped with miRNA targets were selected for ccRCC miRNA-DEG target regulatory network construction. Green – downregulated genes; red – upregulated genes; yellow – miRNAs. ccRCC – clear cell renal cell carcinoma; miRNA – microRNA; DEGs – differentially expressed genes. 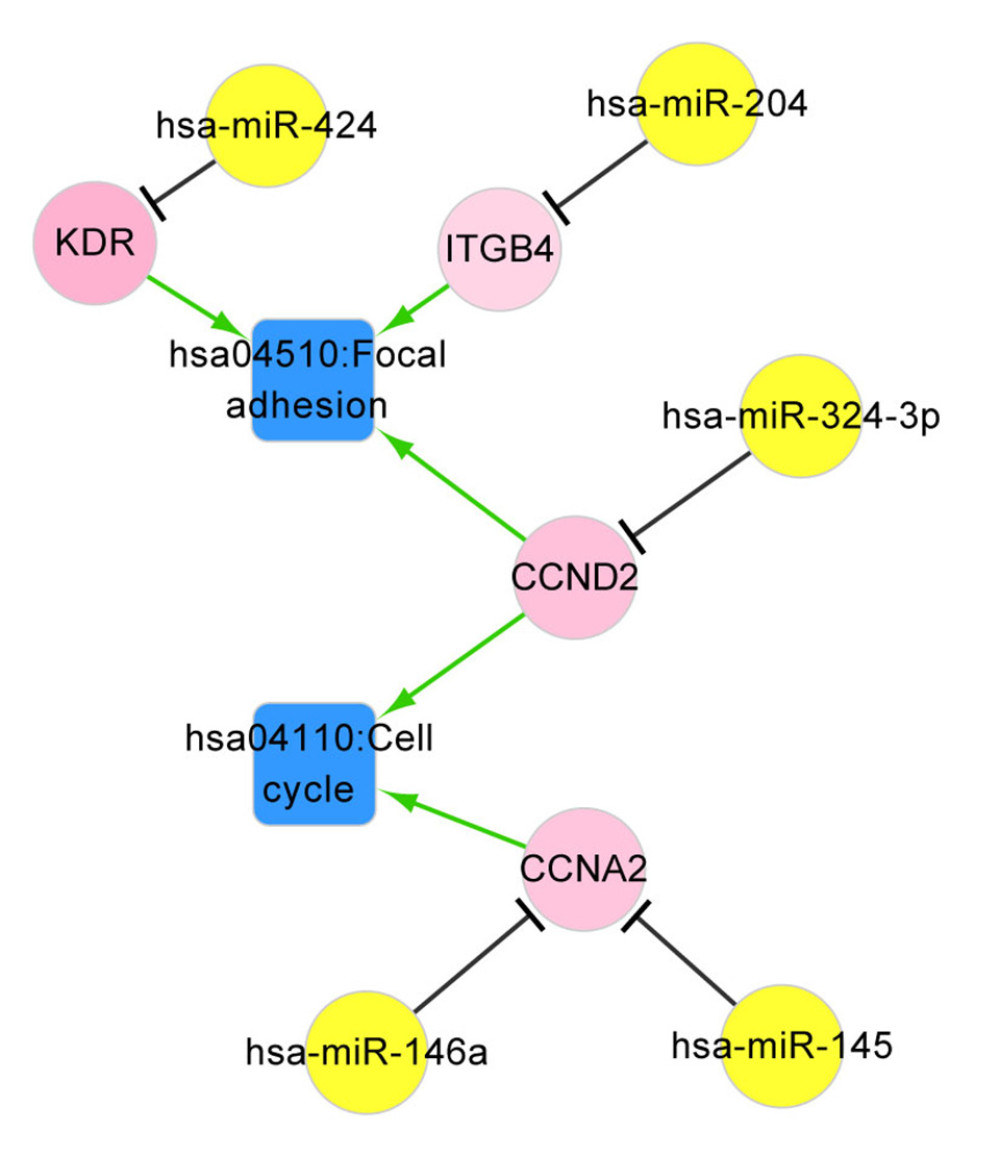 Figure 5. The ccRCC associated pathway network. The pathways closely related with ccRCC were retrieved from the Comparative Toxicogenomics Database, among which the pathways overlapped with those enriched by miRNA targets were used for pathway network constructed. Red – upregulated genes; yellow – miRNAs; blue – ccRCC associated pathways. ccRCC – clear cell renal cell carcinoma; miRNA – microRNA.
Figure 5. The ccRCC associated pathway network. The pathways closely related with ccRCC were retrieved from the Comparative Toxicogenomics Database, among which the pathways overlapped with those enriched by miRNA targets were used for pathway network constructed. Red – upregulated genes; yellow – miRNAs; blue – ccRCC associated pathways. ccRCC – clear cell renal cell carcinoma; miRNA – microRNA. References
1. Hsieh JJ, Purdue MP, Signoretti S, Renal cell carcinoma: Nat Rev Dis Primers, 2017; 3; 17009
2. Sauk SC, Hsu MS, Margolis DJA, Clear cell renal cell carcinoma: Multiphasic multidetector CT imaging features help predict genetic karyotypes: Radiology, 2011; 261; 854-62
3. Garcia JG, Picken MM, Flanigan RC, The importance of histology and cytogenetics in decision making for renal cell carcinoma: World J Urol, 2008; 26; 155-60
4. Patard JJ, Leray E, Riouxleclercq N, Prognostic value of histologic subtypes in renal cell carcinoma: A multicenter experience: J Cli Oncol, 2005; 175; 481-82
5. Golub TR, Slonim DK, Tamayo P, Molecular classification of cancer: Class discovery and class prediction by gene expression monitoring: Science, 1999; 286(5439); 531-37
6. King HC, Sinha AA, Gene expression profile analysis by DNA microarrays: Promise and pitfalls: JAMA, 2001; 286; 2280-88
7. Apostolova LG, Di LJ, Duffy EL, Risk factors for behavioral abnormalities in mild cognitive impairment and mild Alzheimer’s disease: Dement Geria Tr Cogn, 2014; 37; 315-26
8. The Cancer Genome Atlas Research Network, Comprehensive molecular characterization of clear cell renal cell carcinoma: Nature, 2013; 499; 43-49
9. Majmundar AJ, Wong WJ, Simon MC, Hypoxia-inducible factors and the response to hypoxic stress: Mol Cell, 2010; 40; 294-309
10. Semenza GL, HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations: J Clin Invest, 2013; 123; 3664-71
11. Hakimi AA, Reznik E, Lee C-H, An integrated metabolic atlas of clear cell renal cell carcinoma: Cancer Cell, 2016; 29; 104-16
12. Escudier B, Porta C, Schmidinger M, Renal cell carcinoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up: Ann Oncol, 2016; 27; v58-68
13. Tian ZH, Yuan C, Yang K, Gao XL, Systematic identification of key genes and pathways in clear cell renal cell carcinoma on bioinformatics analysis: Ann Transl Med, 2019; 7; 18
14. Yang H, Li W, Lv Y, Exploring the mechanism of clear cell renal cell carcinoma metastasis and key genes based on multi-tool joint analysis: Gene, 2019; 720; 3
15. Khan J, Saal LH, Bittner ML, Expression profiling in cancer using cDNA microarrays: Electrophoresis, 2015; 20; 223-29
16. Yang X, Sun X, Meta-analysis of cancer gene-profiling data: Met Mol Biol, 2010; 576; 409
17. Clough E, Barrett T, The Gene Expression Omnibus Database: Methods Mol Biol, 2016; 1418; 93-110
18. Gumz ML, Zou H, Kreinest PA, Secreted frizzled-related protein 1 loss contributes to tumor phenotype of clear cell renal cell carcinoma: Clin Cancer Res, 2007; 13; 4740-49
19. Tun HW, Marlow LA, von Roemeling CA, Pathway signature and cellular differentiation in clear cell renal cell carcinoma: PLoS One, 2010; 5; 0010696
20. Lenburg ME, Liou LS, Gerry NP, Previously unidentified changes in renal cell carcinoma gene expression identified by parametric analysis of microarray data: BMC Cancer, 2003; 3; 31
21. Gerlinger M, Horswell S, Larkin J, Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing: Nat Genet, 2014; 46; 225-33
22. Parrish RS, Spencer HJ, Effect of normalization on significance testing for oligonucleotide microarrays: J Biopharm Stat, 2004; 14; 575-89
23. Chang L-C, Lin H-M, Sibille E, Tseng GC, Meta-analysis methods for combining multiple expression profiles: Comparisons, statistical characterization and an application guideline: BMC Bioinformatics, 2013; 14; 368
24. Huang DW, Sherman BT, Lempicki RA, Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources: Nat Protocol, 2008; 4; 44
25. Roth A, Szklarczyk D, Simonovic M, The STRING database in 2017: Quality-controlled protein–protein association networks, made broadly accessible: Nucleic Acids Res, 2016; 45; D362-68
26. Shannon P, Markiel A, Ozier O, Cytoscape: A software environment for integrated models of biomolecular interaction networks: Genome Res, 2003; 13; 2498-504
27. Barthélemy M, Betweenness centrality in large complex networks: The European Physical Journal B, 2004; 38; 163-68
28. Ramana J, RCDB: Renal cancer gene database: BMC Res Not, 2012; 5; 246
29. Dweep H, Gretz N, MiRWalk2.0: A comprehensive atlas of microRNA-target interactions: Nat Methods, 2015; 12; 697
30. Grondin CJ, Sciaky D, Wiegers J, The comparative toxicogenomics database: update 2019: Nucleic Acids Res, 2018; 47; D948-54
31. Atrih A, Mudaliar MAV, Zakikhani P, Quantitative proteomics in resected renal cancer tissue for biomarker discovery and profiling: Brit J Cancer, 2014; 110; 1622-33
32. Luo Y, Chen L, Wang G, PPARalpha gene is a diagnostic and prognostic biomarker in clear cell renal cell carcinoma by integrated bioinformatics analysis: J Cancer, 2019; 10; 2319-31
33. Finocchiaro G, Mancuso F, Muller H, Mining published lists of cancer related microarray experiments: Identification of a gene expression signature having a critical role in cell-cycle control: BMC Bioinformatics, 2005; 6(Suppl 4); S14
34. Struckmann K, Schraml P, Simon R, Impaired expression of the cell cycle regulator BTG2 is common in clear cell renal cell carcinoma: Cancer Res, 2004; 64(5); 1632-38
35. Menashe I, Yuenger J, Yeager M, Pathway-based evaluation of 380 candidate genes and lung cancer susceptibility suggests the importance of the cell cycle pathway: Carcinogenesis, 2008; 29; 1938-43
36. Büschges R, Weber RG, Actor B, Amplification and expression of cyclin D genes (CCND1 CCND2 and CCND3) in human malignant gliomas: Brain Pathol, 1999; 9; 435-42
37. Li L, Sarver AL, Alamgir S, Subramanian S, Downregulation of microRNAs miR-1, -206 and -29 stabilizes PAX3 and CCND2 expression in rhabdomyosarcoma: Lab Invest, 2012; 92; 571
38. Li Y-L, Wang J, Zhang C-Y, MiR-146a-5p inhibits cell proliferation and cell cycle progression in NSCLC cell lines by targeting CCND1 and CCND2: Oncotarget, 2016; 7; 59287-98
39. Luo T, Chen X, Zeng S, Bioinformatic identification of key genes and analysis of prognostic values in clear cell renal cell carcinoma: Oncol Lett, 2018; 16; 1747-57
40. Zhu C, Shao P, Bao M, miR-154 inhibits prostate cancer cell proliferation by targeting CCND2: Urol Oncol, 2014; 32; 31.e9-16
41. Petillo D, Kort EJ, Anema J, MicroRNA profiling of human kidney cancer subtypes: Int J Oncol, 2009; 35; 109-14
42. Cho RJ, Huang M, Campbell MJ, Transcriptional regulation and function during the human cell cycle: Nat Genet, 2001; 27; 48-54
43. Zhang X, Ye ZH, Liang HW, Down-regulation of miR-146a-5p and its potential targets in hepatocellular carcinoma validated by aTCGA- andGEO-based study: Febs Open Bio, 2017; 7; 504-21
44. Liang W, Hui T, Venugopal T, Gene networks and microRNAs implicated in aggressive prostate cancer: Cancer Res, 2015; 69; 9490-97
45. Ozen M, Karatas OF, Gulluoglu S, Overexpression of miR-145–5p inhibits proliferation of prostate cancer cells and reduces SOX2 expression: Cancer Inves, 2015; 33; 251-58
46. McLean GW, Carragher NO, Avizienyte E, The role of focal-adhesion kinase in cancer – a new therapeutic opportunity: Nat Rev Cancer, 2005; 5; 505-15
47. Jeong KY, Inhibiting focal adhesion kinase: A potential target for enhancing therapeutic efficacy in colorectal cancer therapy: World J Gastrointest Oncol, 2018; 10(10); 290-92
48. Sato Y, Kanno S, Oda N, Properties of two VEGF receptors, Flt-1 and KDR, in signal transduction: Ann NY Acad Sci, 2010; 902; 201-7
49. Ferraro A, Kontos CK, Boni T, Epigenetic regulation of miR-21 in colorectal cancer: ITGB4 as a novel miR-21 target and a three-gene network (miR-21-ITGB4-PDCD4) as predictor of metastatic tumor potential: Epigenetics, 2014; 9; 129-41
50. Stemmer K, Ellinger-Ziegelbauer H, Ahr H-J, Dietrich DR, Molecular characterization of preneoplastic lesions provides insight on the development of renal tumors: Am J Pathol, 2009; 175; 1686-98
Figures
Figure 1. The significant GO function and pathways for downregualted genes (A) and upregulated genes (B). The downregulated and upregulated genes were subjected to GO function and pathway enrichment analysis by Fisher’s exact test. Red – KEGG pathway; yellow – molecular function; green – cell component; blue – biological process. GO – gene ontology; KEGG – Kyoto Encyclopedia of Genes and Genomes.Figure 2. PPI network. The differentially expressed genes were mapped to proteins based on STRING database. PPI network was visualized using Cytoscape software. Green – downregulated genes; red – upregulated genes. The size of node indicates the node degree. PPI – protein–protein interaction; STRING – Search Tool for the Retrieval of Interacting Genes/Proteins.Figure 3. Modules in PPI network. The modules in PPI network were analyzed using Cytoscape plugin Molecular Complex Detection (Mcode). With cutoff degree ≥2, cutoff node score ≥0.2 and K-core ≥2, 6 modules were obtained. Green – downregulated genes; red – upregulated genes. The size of node indicates the node degree. PPI – protein–protein interaction.Figure 4. The ccRCC associated miRNA regulatory network. The ccRCC-related miRNAs were retrieved from the Renal Cancer Gene Database and DEGs that overlapped with miRNA targets were selected for ccRCC miRNA-DEG target regulatory network construction. Green – downregulated genes; red – upregulated genes; yellow – miRNAs. ccRCC – clear cell renal cell carcinoma; miRNA – microRNA; DEGs – differentially expressed genes.Figure 5. The ccRCC associated pathway network. The pathways closely related with ccRCC were retrieved from the Comparative Toxicogenomics Database, among which the pathways overlapped with those enriched by miRNA targets were used for pathway network constructed. Red – upregulated genes; yellow – miRNAs; blue – ccRCC associated pathways. ccRCC – clear cell renal cell carcinoma; miRNA – microRNA. Tables
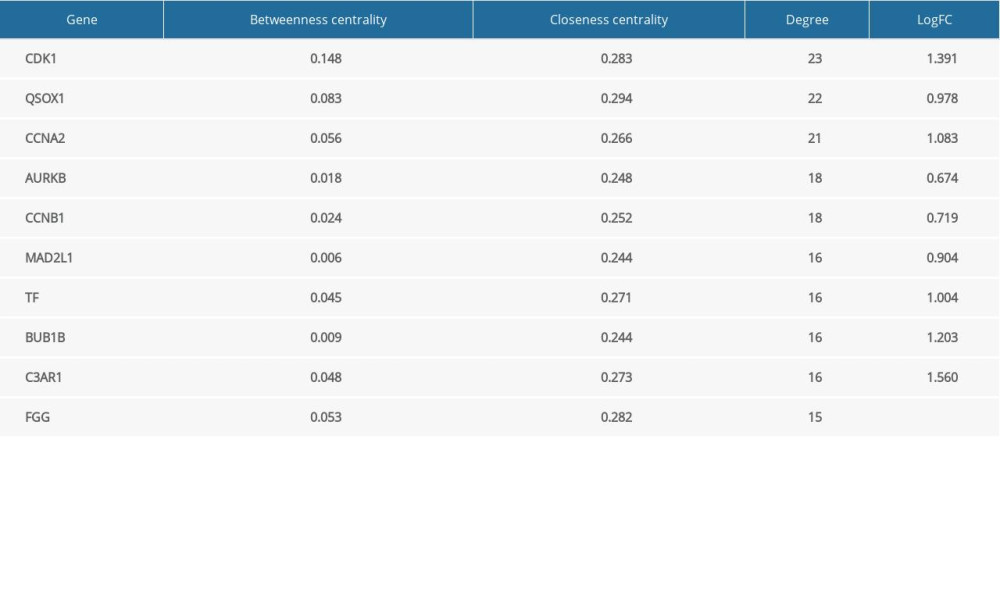 Table 1. Top 10 hub genes based on netweenness centrlity, closeness centrality and degree.
Table 1. Top 10 hub genes based on netweenness centrlity, closeness centrality and degree.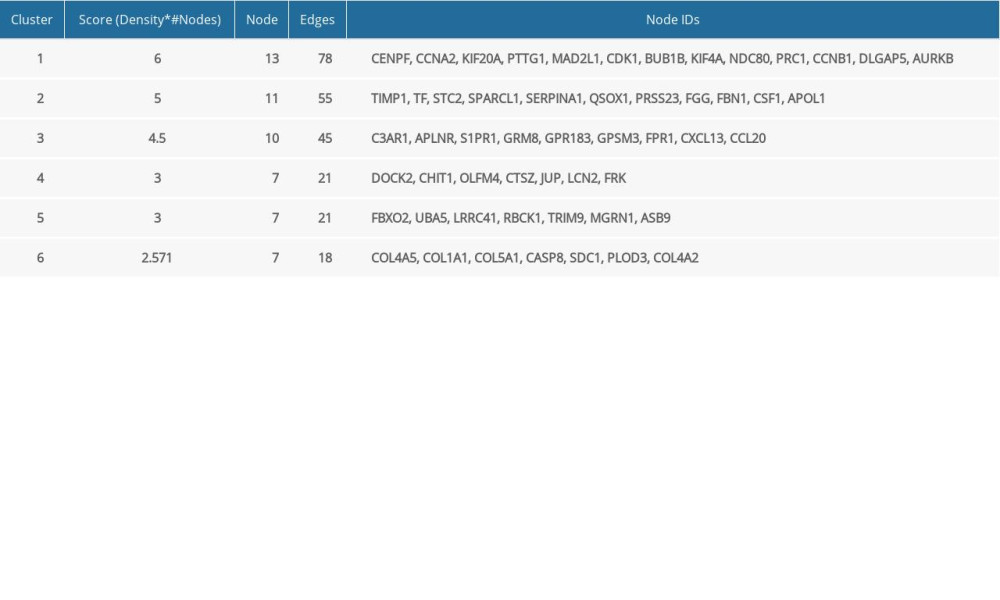 Table 2. Detailed information of modules in PPI network.
Table 2. Detailed information of modules in PPI network.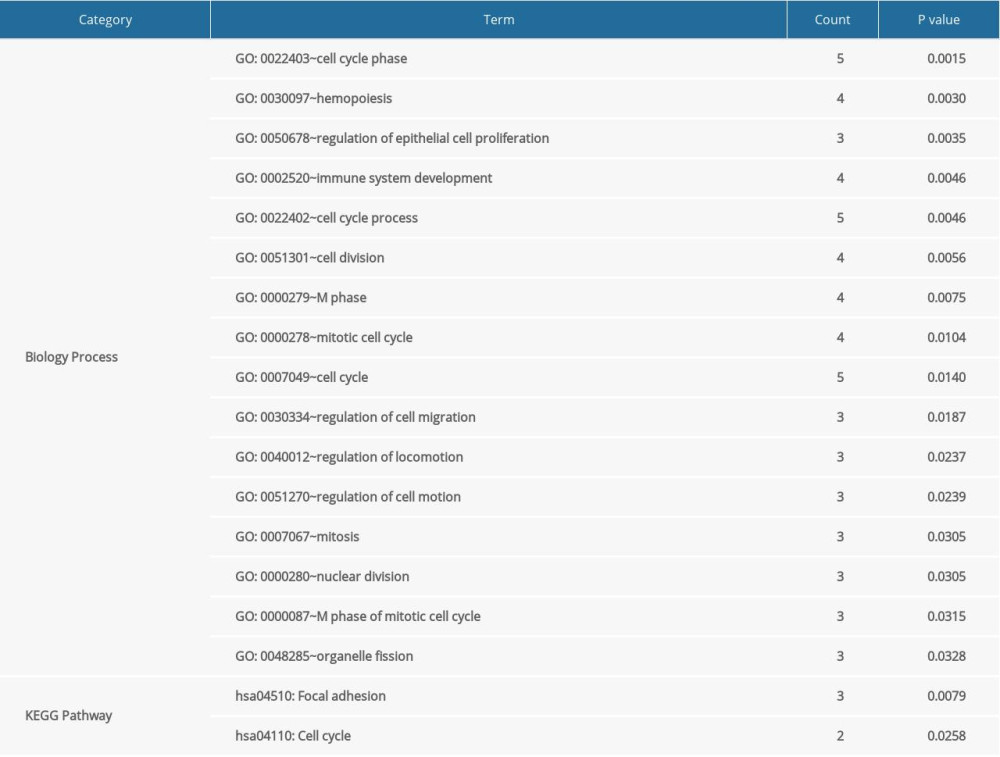 Table 3. Signficant GO functions and pathways for DEGs in miRNA regulatory network.Table 1. Top 10 hub genes based on netweenness centrlity, closeness centrality and degree.Table 2. Detailed information of modules in PPI network.Table 3. Signficant GO functions and pathways for DEGs in miRNA regulatory network.
Table 3. Signficant GO functions and pathways for DEGs in miRNA regulatory network.Table 1. Top 10 hub genes based on netweenness centrlity, closeness centrality and degree.Table 2. Detailed information of modules in PPI network.Table 3. Signficant GO functions and pathways for DEGs in miRNA regulatory network. In Press
Clinical Research
Effects of Single-Bout Endurance Exercise Intensity on Peripheral Neurotrophic Factors in Patients With Isc...Med Sci Monit In Press; DOI: 10.12659/MSM.952089
Review article
Anisodus tanguticus in Cancer Research: A Review of Traditional Use, Phytochemistry, Extraction Methods, an...Med Sci Monit In Press; DOI: 10.12659/MSM.952999
Clinical Research
Nasal Mucociliary Clearance and Its Relationship With Disease Severity in Patients With Multiple SclerosisMed Sci Monit In Press; DOI: 10.12659/MSM.952850
Clinical Research
Modified Thoracoabdominal Nerves Block Through the Perichondrial Approach vs Subcostal Transversus Abdomini...Med Sci Monit In Press; DOI: 10.12659/MSM.953976
Most Viewed Current Articles
17 Jan 2024 : Review article 14,176,570
Vaccination Guidelines for Pregnant Women: Addressing COVID-19 and the Omicron VariantDOI :10.12659/MSM.942799
Med Sci Monit 2024; 30:e942799
13 Nov 2021 : Clinical Research 3,762,188
Acceptance of COVID-19 Vaccination and Its Associated Factors Among Cancer Patients Attending the Oncology ...DOI :10.12659/MSM.932788
Med Sci Monit 2021; 27:e932788
14 Dec 2022 : Clinical Research 2,466,310
Prevalence and Variability of Allergen-Specific Immunoglobulin E in Patients with Elevated Tryptase LevelsDOI :10.12659/MSM.937990
Med Sci Monit 2022; 28:e937990
16 May 2023 : Clinical Research 708,927
Electrophysiological Testing for an Auditory Processing Disorder and Reading Performance in 54 School Stude...DOI :10.12659/MSM.940387
Med Sci Monit 2023; 29:e940387