02 July 2020: Clinical Research
A Seven-Gene Signature with Close Immune Correlation Was Identified for Survival Prediction of Lung Adenocarcinoma
Xuan Zou1ABCDEF, Zhihuang Hu1AB, Changjing Huang1DF, Jianhua Chang1AG*DOI: 10.12659/MSM.924269
Med Sci Monit 2020; 26:e924269






Abstract
BACKGROUND: Lung adenocarcinoma is the most life-threatening malignancy with high incidence and poor long-term survival. The crucial role of tumor immunity reveals the significance of exploring immune-related prognostic predictors in lung adenocarcinoma.
MATERIAL AND METHODS: Immune-related genes were screened out applying the ESTIMATE algorithm. The Cancer Genome Atlas Lung Adenocarcinoma (TCGA-LUAD) dataset was trained for the construction of Cox proportional hazard model. Univariate Cox and Lasso regression analysis was conducted to reduce the overfitting of model. Nomogram integrated the signature and clinicopathological characteristics was established for prognosis prediction of LUAD. The GSE30219, GSE41271, and GSE42127 datasets were analyzed for external validation. LUAD patients were separated into low-risk and high-risk subgroups based on the optimum cutoff threshold of calculated risk score. The predictive value of the signature was evaluated using Kaplan-Meier survival analysis, Harrell’s C-index, and receiver operating characteristic (ROC) curve analysis and calibration curve. Clinical- and immune-correlation of the signature was further performed. Gene Set Enrichment Analysis (GSEA) was performed for functional exploration.
RESULTS: An immune-related signature containing 7 genes was identified. The signature exhibited reliable performance in the prediction of overall survival for LUAD with the C-index being 0.72. The areas under the curve (AUCs) of the model in 1-year risk prediction were 0.781, 0.797, 0.659, and 0.822 for TCGA-LUAD, GSE30219, GSE41271, and GSE42127 datasets, respectively. In all datasets, the signature proved to an independent risk factor for LUAD. Correlation analyses and GSEA further revealed the close relationship between the predictive biomarker and tumor immunity.
CONCLUSIONS: A Cox proportional hazard model consisting of 7 genes was identified for prognostic prediction of LUAD. The signature was highly correlated with immunity and deserves further exploration.
Keywords: nomograms, Adenocarcinoma of Lung, Aged, 80 and over, Area Under Curve, Biomarkers, Tumor, Databases, Genetic, Multivariate Analysis, Proportional Hazards Models, ROC Curve, Risk Factors
Background
Lung cancer (LC), ranking as one of the top 3 in incidence and mortality rate, is the most prevalently diagnosed cancers worldwide both for males and females. Though the morbidity has decreased gradually in many regions due to effective tobacco control and health management measures, LC is still the one of the main causes of cancer-associated death and has worse long-term survival compared with many other cancers [1]. Lung adenocarcinoma (LUAD) is the main histopathology type of non-small cell lung cancer (NSCLC), with a proportion of around 40% of all LC cases [2]. The development of traditional treating strategies including surgery, chemotherapy, radiotherapy, and targeted therapy has helped improve the clinical outcome of LUAD patients [2]. However, with an overall 5-year survival rate less than 15%, the clinical prognosis of LUAD remains unsatisfactory, especially for patients with an advanced disease stage [3,4]. Previous research has identified several recognized prognostic factors for LUAD such as age, TNM stage, smoking history, and gene mutation status [5–8]. Several molecules such as p16 and HIF not only have profound influence on cancer development but also act as prognostic indicators for lung cancer according to previous studies [9,10]. A growing number of risk stratification models which combine numbers of parameters have also been proposed for prognostic prediction of LUAD. However, the capacity to define high-risk LUAD patients and improve their future outcomes still remains limited in clinical practice.
With the deepening understanding of immune dysregulation in cancer development, the treatment of LC has entered a new era of immunotherapy. Accumulating evidence has revealed that immune-related mechanisms may have profound impact on NSCLC survival. Interactions between immune cells, cytokines, stromal components, and cancer cells can regulate the pro- or anti-tumor activity of tumor microenvironment (TME), which affect the survival of NSCLC patients [11–13]. Moreover, the use of some concomitant medications of immune checkpoint inhibitors might also have profound impact on the clinical outcome of NSCLC patients [14]. Identification of immune-related prognostic markers not only assists in risk definition and treatment decision, but also provides inspiration for future mechanism exploration. However, research focused on the discovery of immune-related prognostic signatures in LUAD is still inadequate and lack in accurate verification up to now.
Currently, rapid advances in high-throughput technologies and bioinformatics methodology have offered a brand-new avenue for precise oncology research. Among the present cancer genomics programs, The Cancer Genome Atlas (TCGA) is the largest and richest, which integrates genomic, epigenomic, transcriptomic, and proteomic data of 33 common cancer types. Gene Expression Omnibus (GEO) is a public database providing abundant raw and processed high-throughput data generated in different platforms [15]. Integrative analysis of TCGA and GEO data may provide more insight into precise investigation. In addition, several new algorithms such as ESTIMATE and CIBERSORT enable researchers to interpret gene expression data into TME and tumor-infiltrating lymphocytes (TILs) information, which greatly facilitates research on tumor immunity [16,17].
In this study, we calculated the degree of immune infiltration in samples of TCGA-LUAD by using the ESTIMATE method. Survival analysis confirmed the potential association between immune components and overall survival in LUAD patients. A prognostic model containing 7 genes were then built by applying Cox and Lasso regression analyses on the basis of immune relevant genes. The identified prognostic biomarker was further validated in 3 external GEO datasets: GSE30219, GSE41271, and GSE42127. We aimed to construct an immune-related prognostic model with high reliability in risk prediction for LUAD patients. Correlation analysis between the identified signature and immune status of LUAD samples was also performed for functional exploration.
Material and Methods
DATA SOURCE AND PREPARATION:
The TCGA-LUAD dataset which included pathologically confirmed LUAD patients was chosen as the training set for model construction. FPKM-normalized transcriptome profiling data of LUAD tumor tissues were downloaded using the gdc-client tool (
Three independent Gene Expression Omnibus (GEO) datasets (GSE30219, GSE41271, and GSE42127) with both survival information of LUAD patients and high-throughput transcriptome profiling data of LUAD tumor tissues were analyzed for external validation of the identified prognostic signature. All the patients in the 3 sets were pathologically diagnosed as LUAD. The series matrix file and the corresponding platform annotation file (GPL570 for GSE30219, GPL6884 for GSE41271 and GSE42127) of each gene expression microarray dataset were downloaded from
IDENTIFICATION OF IMMUNE-RELATED GENES:
The immune status of TCGA-LUAD samples was inferred by the “estimation of stromal and immune cells in malignant tumor using expression data” (ESTIMATE) method. Briefly, the scores that represent the proportion of infiltrating immune cells (immune score) and stromal cells (stromal score) in TME were output by performing single sample GSEA (ssGSEA). Immune score and stromal score were combined as the ESTIMATE score which predicts the purity of tumor samples. All the TCGA-LUAD samples were divided into high and low immune infiltration group according to the median value of immune scores. Samples with immune scores higher than the median were divided into the high immune infiltration group, while those with lower immune score than the median were divided into the low immune infiltration group. Kaplan-Meier survival analysis comparing the 2 subgroups was performed to assess the relationship between prognosis and immune status in LUAD. Differentially-expressed genes (DEGs) between the 2 groups were further identified using the “limma” R package [18], with the threshold of false discovery rate (FDR) adjusted P-value <0.05 and fold change (FC) >2 or <0.5. Spearman’s rank correlation analysis was further performed to evaluate the correlation between immune scores and the identified genes. Genes with an average FPKM <1 were excluded from model construction due to the extremely limited abundance in tissues.
BUILDING AND VALIDATION OF THE PROGNOSIS PREDICTIVE MODEL:
The TCGA-LUAD dataset was used as the training set for the development of Cox regression model predicting survival in LUAD patients, and 3 GEO datasets (GSE30219, GSE41271, and GSE42127) were explored for external validation. Firstly, univariate Cox regression analysis were conducted for all the identified genes to screen out the prognosis-associated factors with a threshold of P-value <0.01. The LASSO regression analysis was further conducted using the “glmnet” R package [19,20] to reduce the model complexity and multi-collinearity. Variables selected were further filtered using stepwise multi-variate Cox regression method for the construction of optimum model. The predictive risk score of each sample in the 4 datasets was calculated according to the estimated coefficients of variables in the Cox regression model. Patients were separated 2 subgroups (low-risk versus high-risk) according to the optimal cutoff value decided by the X-tile software [21]. Briefly, the file containing the calculated risk score, survival status, and overall survival time of each sample was uploaded to the software (version 3.4.7). The software enumerated all potential cutoff-points that define high- and low-risk patients, and the result of Kaplan-Meier survival analysis for each point was further compared. The point that can most significantly separate the survival curves of the 2 groups was considered to be the optimal cutoff value. The predictive value of the signature was assessed by Kaplan-Meier survival analysis and survival receiver operating characteristic (ROC) curve analysis in the 4 sets, respectively. Calibration curve and the Harrell’s concordance index (C-index) calculated using a 1000-resampling bootstrap method was applied to measure the accuracy and reliability of the identified model. A nomogram combing risk score and several clinical parameters was developed for LUAD survival prediction. In addition, univariate and multivariate Cox regression analyses were further conducted to assess whether the risk score was an independent prognostic variate for LUAD.
IMMUNE CORRELATION ANALYSIS:
The status of tumor microenvironment, immune cell infiltration, tumor mutation burden (TMB), gene expression of immune checkpoints, and human leukocyte antigen (HLA) system was further compared between the 2 subgroups (low-risk versus high-risk) of LUAD patients in the TCGA-LUAD dataset using Mann-Whitney U test. The correlation between risk score and ESTIMATE-generated scores was evaluated using the Spearman’s rank correlation analysis. The CIBERSORT method was applied to enumerate 22 mature tumor infiltration leukocytes (TILs) based on the transcriptome expression data [16]. A P-value was calculated for the global deconvolution of each sample, and only samples with P-value <0.05 were included for analysis. The TMB value of each sample was calculated as previously described [22].
GENE SET ENRICHMENT ANALYSIS (GSEA):
GSEA analysis was conducted to explore the biological processes potentially involved by the prognostic biomarker. Genome-wide expression profiles of all the 19 658 protein-coding genes from TCGA samples belonging to the low-risk and high-risk subgroups were analyzed in GSEA v4.0.3. Four classical gene sets in Molecular Signatures Database including “c2.cp.kegg.v7.0.symbols.gmt (Curated)”, “c5.bp.v7.0 symbols.gmt (Gene Ontology)”, “c5.cc.v7.0 symbols.gmt (Gene Ontology)”, and “c5.mf.v7.0 symbols.gmt (Gene Ontology)” were considered. A normalized enrichment score (NES) in the gene set of interest was calculated, and an FDR-normalized q-value <0.05 was regarded to be of statistical significance.
STATISTICAL ANALYSIS:
R version 3.5.3 (
Results
STUDY DESIGN AND SUBJECTS:
The flow chart of study design is shown in Figure 1. A total of 499 LUAD samples with follow-up time more than 30 days in the TCGA-LUAD dataset were analyzed for the training of the prognostic signature. The association between the identified model and immune status was further explored in the TCGA dataset. Three GEO datasets of GSE30219, GSE41271, and GSE42127 were analyzed for the external validation of the prognostic model. The clinicopathologic characteristics of the 4 independent cohorts are listed in Table 1.
IDENTIFICATION OF IMMUNE-RELATED GENES:
In the TCGA cohort, the ratio of immune and stromal components in tumor tissues was calculated using the ESTIMATE algorithm. All the cases were divided into 2 groups by the median immune score. The result of Kaplan-Meier survival analysis showed that the high-immune score group had significantly better survival than the low-immune score group (P=0.025; Supplementary Figure 1), which indicated the close relationship between immune status and survival in LUAD patients. On the contrary, there was no significant difference in patient survival between the high- and low-stromal score group. The difference of gene expression was further analyzed between the high- and the low-immune score group to identify potential immune-related genes for model construction. A total of 524 dysregulated genes were identified with log2FC >1 and P<0.05 (Supplementary Figure 2, Supplementary Table 1).
CONSTRUCTION OF THE PROGNOSTIC MODEL CONTAINING 7 GENES IN THE TRAINING SET:
Among the 524 immune-related genes, a total of 38 genes that were significantly correlated with the overall survival in LUAD patients were identified (P<0.01). The hazard ratio (HR) and the 95% confidence interval (CI) of each variable are shown in Table 2. The candidate list was further reduced to 16 genes by the Lasso regression method to alleviate model overfitting (Supplementary Figure 3). The following stepwise variable selection procedure finally established an optimal Cox proportional hazard model containing 7 key genes: CIITA (class II major histocompatibility complex transactivator), CLEC7A (C-type lectin domain containing 7A), BIRC3 (baculoviral IAP repeat containing 3), TESC (tescalcin), MCOLN2 (mucolipin 2), DEPDC7 (DEP domain containing 7), and VEGFD (vascular endothelial growth factor-D), see Table 3. The predicted risk score can be calculated with the formula: −0.198926091*CLEC7A +−0.240572316*CIITA +0.360229039*BIRC3+0.318599709*DEPDC7 +0.139659504*TESC +−0.17303622*MCOLN2 + −0.253980504*VEGFD. The samples of the training set were separated into high-risk and low-risk subgroups according to the optimum cutoff threshold of 0.463 calculated by X-tile software. The Kaplan-Meier survival curve showed the significantly worse survival in the high-risk group with P<0.0001 (Figure 2A). The C-index for the biomarker was 0.72 (95% CI: 0.693–0.747), and the calibration curve further showed the consistency between the predicted and actual survival outcomes of the 499 patients (Figure 2B, Supplementary Figure 4). Time-dependent survival ROC curves was constructed to assess the prognostic prediction value of the model. At each time point of 1-year, 2-years, and 3-years, the predictive capability of the biomarker remained well with the AUCs being 0.781, 0.664, and 0.652, respectively. The risk distribution plot and heat-map of gene expression in the training set are shown in Figure 2C–2E. When several clinical features including age, gender, and TNM stage were combined into analysis, the multiple ROC curves indicated that the risk score could outperform other factors with higher AUCs in outcome prediction of LUAD patients (Figure 3A). Multivariate Cox regression analysis revealed that the risk score could independently predict the clinical outcome in LUAD with P<0.001 (Figure 3B). A prognostic nomogram incorporating risk score and several clinical characteristics was developed to predict overall survival in LUAD patients (Figure 3C).
EXTERNAL VALIDATION IN GEO DATASETS:
The signature’s predictive performance was further validated in 3 independent GEO datasets (GSE30219, GSE41271, and GSE42127). The risk score of each patient in the testing sets was calculated according to the relative expression levels of the 7 genes, using the same formula established in the training set of TCGA-LUAD. In each dataset, samples were separated into low-risk and high-risk groups with the same cutoff value. The Kaplan-Meier curves generated from the 3 external datasets showed consistently significant difference in survival status between high- and low-risk patients with P<0.05 (Figure 4A, 4D, 4G). Patients with high risk had markedly worse survival outcomes than patients with low risk. The AUCs of the 7-gene signature in predicting 1-year survival of LUAD patients were 0.797 in GSE30219, 0.659 in GSE41271, and 0.822 in GSE42127 (Figure 4B, 4E, 4H, and Supplementary Figure 5 show the results of time-dependent ROC curves). In GSE42127 dataset, the identified model had much better performance with higher AUC than the TNM stage in prognostic prediction. Multivariate Cox regression analysis verified that the risk score was an independent prognostic variate in all the 3 testing datasets with P<0.05 and HR >1 (Figure 4C, 4F, 4I). The reliability of the 7-gene prognostic biomarker was proved well in the testing sets.
CORRELATION ANALYSIS BETWEEN RISK SCORE AND CLINICOPATHOLOGIC FEATURES:
Risk scores were compared between subgroups with different clinicopathologic features in the TCGA-LUAD cohort. As shown in Figure 5, patients with the following characteristics had significantly higher risk score than the corresponding control groups with P<0.05: i) males (versus females); ii) advanced TNM stage (stage III/IV versus stage I/II); iii) larger tumor size (T3/T4 versus T1/T2); and iv) lymph node invasion (N1/N2/N3 versus N0). Moreover, it was observed that the risk scores of KRAS-mutant patients were much higher than KRAS wild-type patients. The similar disparity was not found in EGFR (epidermal growth factor receptor) and ALK (anaplastic large-cell lymphoma kinase) mutations.
IMMUNE-RELEVANCE ANALYSIS OF THE PROGNOSTIC MODEL IN TCGA DATASET:
Remarkable difference of immune and stromal fraction in TME was observed between the 2 subgroups in the TCGA-LUAD cohort. Correlation analysis indicated that the risk score predicted by the 7-gene prognostic model was negatively associated with immune score, estimate score, and combined ESTIMATE score with R<−0.4 and P<0.001 (Figure 6A–6C). Higher risk score indicated lower degree of immune and stromal cells infiltration in LUAD tissues. Tumor mutation burden (TMB) levels were further compared between patients with high-risk and low-risk, but no significant difference was discovered (Figure 6D). The gene expression of immune checkpoints (PD-1, PD-L1, PD-L2, and CTLA-4) and HLA molecules in tumor tissues was further analyzed. The high-risk patients had much lowered mRNA expression levels of these immune-related molecules compared with the low-risk patients, which might reflect the alteration of immune status in tumor microenvironment (Figure 6E, 6F). Furthermore, in order to elucidate the precise immune status of tumor samples, 22 types of mature human TILs were quantified using the CIBERSORT method. For samples with meaningful P-values, the estimated proportions of activated natural killer (NK) cells, memory B cells, plasma cells, M0 macrophages, M2 macrophages, monocytes, resting dendritic cells, and resting mast cells significantly varied between the high-risk and low-risk patients, which deserves further exploration (Figure 6G). The results further revealed the mutual relationship between the identified 7-gene signature, immune status, and disease outcomes in LUAD.
GENE SET ENRICHMENT ANALYSIS (GSEA):
GSEA was conducted to compare the difference of molecular mechanisms involved in the high- and low-risk patients in the TCGA-LUAD dataset. Supplementary Table 2 showed the significant 811 pathways enriched in the low-risk patients (FDR <0.05). Notably, most of these pathways or biological processes are closely correlated with immunity, which include “chemokine signaling pathway”, “T cell activation”, “T cell differentiation”, “immune response regulating cell surface receptor signaling pathway”, “adaptive immune response”, “regulation of immune effector process”, etc. Figure 7 list some of these enrichment results. The results showed a strong connection between the identified signature and tumor immunity, which makes sense in both prognostic prediction and treatment decision in LUAD.
Discussion
There has been growing awareness in oncology research that cancer is a complex ecosystem composed of both tumor cells and nontumor cells. Nontumor components in tumor tissues form TME and can impact tumor growth, invasion, metastasis, and prognosis [23,24]. Immune contexture in TME, which is determined by the composition, location, and function of immune cell populations, can transfer information about the characteristics and destinies of many cancer types, including LUAD [25–27]. Though TNM stage system has been traditionally regarded as standard determinant of overall survival in LUAD patients, the features of TILs and immune-related molecules were discovered to be more powerful predictors of clinical outcomes [28]. For example, in LUAD, it was reported that tumor-associated macrophage infiltration may contribute to poor clinical outcomes [29]; the infiltration of NK cells subset CD57+ cells can predict a good prognosis [30]; the expression of HLA class I antigen is a favorable prognostic factor [31]; the upregulation of PD-L1 can lead to worse prognosis, etc. [32]. Several peripheral immune-based biomarkers such as soluble inflammatory factors and circulating T cells were also proposed as the predictive biomarker [33]. However, due to the limitations of testing methods, the detection of some immune-related biomarkers is often unstable or impractical. In addition, prediction based on a single biomarker can be less reliable than a comprehensive model integrating multiple factors. Gene expression signatures, with the advantages of being informative and easily monitored, are thus ideal predictors of cancer prognosis [34].
In this study, we found that patients with higher immune-score had remarkably better clinical outcome than patients with lower immune-score. The result further suggested the close relationship between tumor immunity and clinical outcome in LUAD. A Cox proportional hazard model containing 7 genes (CIITA, CLEC7A, BIRC3, TESC, MCOLN2, DEPDC7, and VEGFD) was constructed for survival prediction of LUAD patients. The risk score derived from the model had good and stable performance in prognostic prediction of LUAD. In both the training (the TCGA-LUAD dataset) and testing sets (the GSE30219, GSE41271, and GSE42127 datasets), the risk score consistently proved to be an independent and reliable prognostic variate in LUAD. In TCGA-LUAD and GSE42127 cohorts, the risk score was discovered to be superior in survival prediction than TNM stage system. Furthermore, a nomogram combining the identified signature and several clinical factors (age, gender, and TNM stage) was built for the exact estimation of patients’ survival probability in LUAD.
The results of immune-correlation analyses and GSEA showed a strong association between the identified signature and tumor immune status. It was found that high-risk patients seemed to have lower immune, stromal fraction, and higher tumor purity in TME. Significant expressing difference of immune checkpoints and HLA genes further indicated the difference of immune status between the high-risk and low-risk LUAD patients. GSEA revealed that the main differences between the 2 groups was largely found in the immune-related biological processes. The analysis of 22 mature TILs further demonstrated the potential association between the identified signature and tumor immune contexture. The exploration preliminarily showed a mutual crosstalk between the 7-gene biomarker, clinical outcomes, and immune characteristics of TME in LUAD patients, which deserves deeper research in the future. Moreover, subgroup analyses showed that the risk scores calculated by the immune-correlated prognostic model were significantly different between KRAS-mutant and KRAS-wild type patients. In NSCLC, mutation in KRAS, EGFR, and ALK were most commonly happened genetic variation types [35]. Unlike other mutation types, NSCLC patients with KRAS mutation response poorly to target therapy and often have worse prognosis. Recently, growing evidence revealed that KRAS-mutant NSCLC cases especially those with concomitant TP53 mutation might benefit from immune checkpoint inhibition therapy [36]. The present study further demonstrated the close relationship between KRAS mutation and cancer immune response in LUAD patients.
Among the 7 genes that built the model, 4 genes (CIITA, CLEC7A, MCOLN2, and VEGFD) were favorable prognostic factors, while 3 genes (BIRC3, DEPDC7, and TESC) were unfavorable prognostic factors for LUAD patients. Up to now, there have been extensive reports about the roles of these genes in tumor immunity and prognosis in NSCLC or other cancer types. CIITA, the master expressing regulator of the major histocompatibility class (MHC) class II molecules and accessory genes for MHCII-restricted antigen presentation, is crucial for normal immune function [37]. Though most tumor cells express MHC class I molecules rather than class II, MHCII-mediated CD4+ T cells activation is critical for host anti-tumor immunity [38]. CIITA deficiency in solid tumors would cause the inability of tumor cells to trigger TH cells [39]. It was once reported that CIITA knockdown could reduce the sensitivity to immunotherapy in NSCLC cells. According to He et al., higher expression of MHC-II molecules was correlated with better survival in NSCLC patients [40]. The trend we observed in our study was consistent with the results generated by previous research. CLEC7A (also known as Dectin-1) is a member of pattern-recognition receptor (PPR) that functions in comprehensive immunomodulation [41]. In TME, CLEC7A critically enhances the tumor cell-killing effect mediated by NK cells [42]. As the most important C-type lectin receptor, CLEC7A is highly involved in comprehensive immune responses, the expression of which could significantly regulate cancer risk [43]. MCOLN2 (mucolipin 2) belongs to the transient receptor potential mucolipin-like family that mainly functions in membrane trafficking events regulation [44]. It is worth noting that unlike its 2 paralogs, MCOLN1 and MCOLN3, MCOLN2 may play an important role in immune response by enhancing viral infection [45]. MCOLN2 expression is also found to be promoted by B-cell lineage specific activator protein PAX5 [46]. The exact effect of MCOLN2 in cancer immunity and survival is still unclear, but our research could be a hint to further exploration. VEGFs are highly engaged in angiogenesis pathway, and VEGF/VEGFR signaling pathway has a complicated crosstalk with immune in TME [47]. According to Carrillo et al., higher expression of VEGFD was significantly associated with the better overall survival for NSCLC patients at advanced stage, which was similar to our findings [48].
Multivariate Cox regression analysis indicated that BIRC3 and DEPDC7 were independent predictors for worse prognosis in LUAD patients. BIRC3 (also known as cIAP2) belongs to inhibitor of apoptosis proteins. The role BIRC3 plays in a variety of cancers has been frequently observed. It was reported that BIRC3 could repress the constitutive activation of NF-κB signaling and regulate the survival of various immune cells, having important function in inflammation and innate immune [49–51]. According to Egildo et al., DEPDC7 could induce the activation of NF-κB which is a curial regulator of immune response [52]. But at the moment, information about the biological function of DEPDC7 is still quite limited. TESC (tescalcin) has been reported as a regulator of cell growth and differentiation, and also involved in the progression of various cancers. In LUAD, upregulated TESC is found to be correlated with enhanced epithelial-mesenchymal transition process and cancer stem cell-like characteristics [53]. The exact roles of these genes in cancer immunity deserves further investigation in the future.
In this study, we constructed a potent prognostic model for the survival prediction of LUAD. The identified 7-gene signature is highly correlated with cancer immunity, delivering underlying information about immunotherapy response and immune-related mechanisms in LUAD. Nonetheless, there were still certain limitations in this study. Firstly, the model was trained and tested using retrospective data, and still required further validation before prospective clinical application. Secondly, there was a considerable amount of missing values such as smoking history and exact TNM classification of patients in the datasets available, which limited the maximum use of statistics. Moreover, the exact molecular mechanisms of these identified genes in cancer immunity and progression still needs further experimental validation.
Conclusions
In summary, we developed an immune-related prognostic model which integrated the expression levels of 7 genes for the prediction of overall survival in LUAD. The signature reliably predicted the clinical outcomes of patients and showed close association with tumor immunity in LUAD.
Supplementary Data
Figures
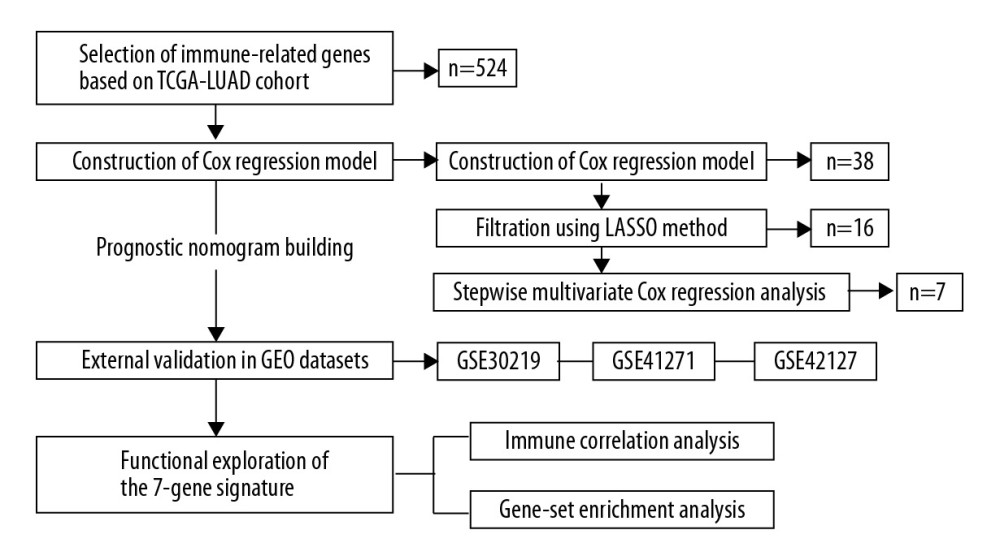 Figure 1. The flow chart of study design.
Figure 1. The flow chart of study design. 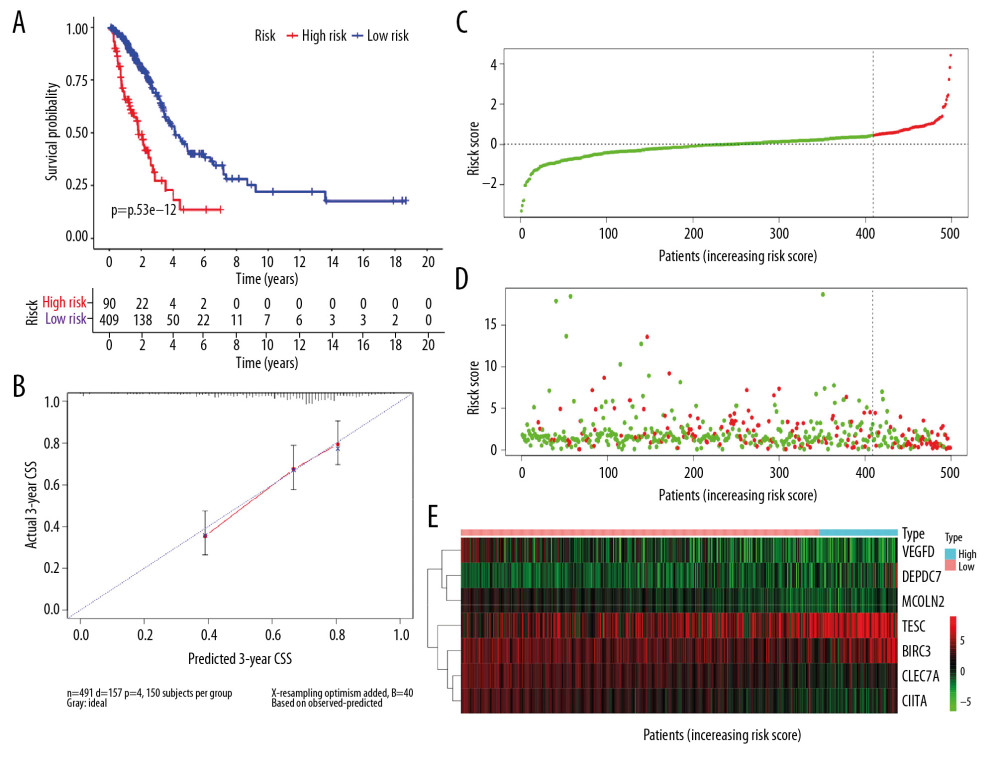 Figure 2. Predicted performance of the identified prognostic signature in the training dataset. (A) The Kaplan-Meier survival analysis of the 7-gene prognostic signature in the TCGA-LUAD cohort. According to the optimal cutoff value, samples are divided into high-risk and low-risk groups. (B) The calibration curves of the model for predicting CSS in LUAD. The predicted 3-year survival probability is plotted on the X-axis; the corresponding actual survival rate is plotted on the Y-axis. (C) The distribution plot of risk score. Patients are arranged from left to right in an increasing order of risk score. (D) The survival status of each patient. Y-axis represents the overall survival time. The color code: green for alive case, red for dead case. (E) The heatmap of mRNA expression levels of the 7 genes. CSS – cancer specific survival; LUAD – lung adenocarcinoma.
Figure 2. Predicted performance of the identified prognostic signature in the training dataset. (A) The Kaplan-Meier survival analysis of the 7-gene prognostic signature in the TCGA-LUAD cohort. According to the optimal cutoff value, samples are divided into high-risk and low-risk groups. (B) The calibration curves of the model for predicting CSS in LUAD. The predicted 3-year survival probability is plotted on the X-axis; the corresponding actual survival rate is plotted on the Y-axis. (C) The distribution plot of risk score. Patients are arranged from left to right in an increasing order of risk score. (D) The survival status of each patient. Y-axis represents the overall survival time. The color code: green for alive case, red for dead case. (E) The heatmap of mRNA expression levels of the 7 genes. CSS – cancer specific survival; LUAD – lung adenocarcinoma. 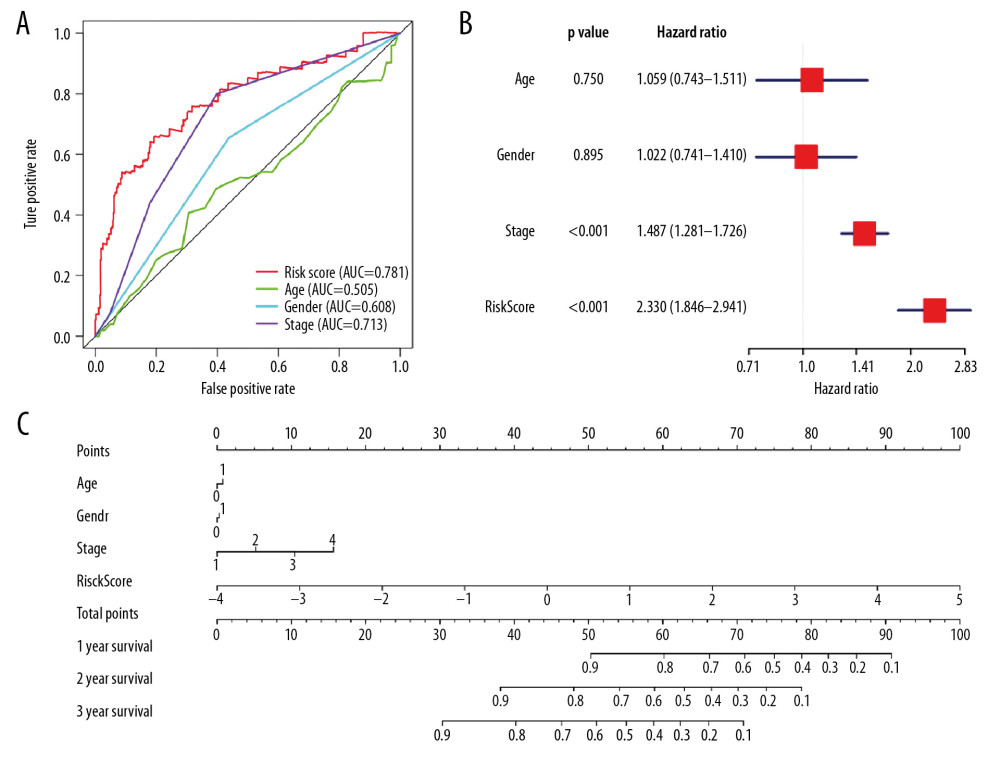 Figure 3. (A) The ROC curves comparing the prognostic values of risk score and several clinical factors in the TCGA-LUAD dataset. The predicted time point is 1 year. ROC curve – receiver operating characteristic curve; AUC – area under the ROC curve; TCGA-LUAD – The Cancer Genome Atlas Lung Adenocarcinoma. (B) The forest plot for multivariate survival analysis based on risk score and other clinical factors in TCGA-LUAD. The hazard ratio and 95% confidence interval are presented. (C) The nomogram constructed for the prediction of survival probability.
Figure 3. (A) The ROC curves comparing the prognostic values of risk score and several clinical factors in the TCGA-LUAD dataset. The predicted time point is 1 year. ROC curve – receiver operating characteristic curve; AUC – area under the ROC curve; TCGA-LUAD – The Cancer Genome Atlas Lung Adenocarcinoma. (B) The forest plot for multivariate survival analysis based on risk score and other clinical factors in TCGA-LUAD. The hazard ratio and 95% confidence interval are presented. (C) The nomogram constructed for the prediction of survival probability. 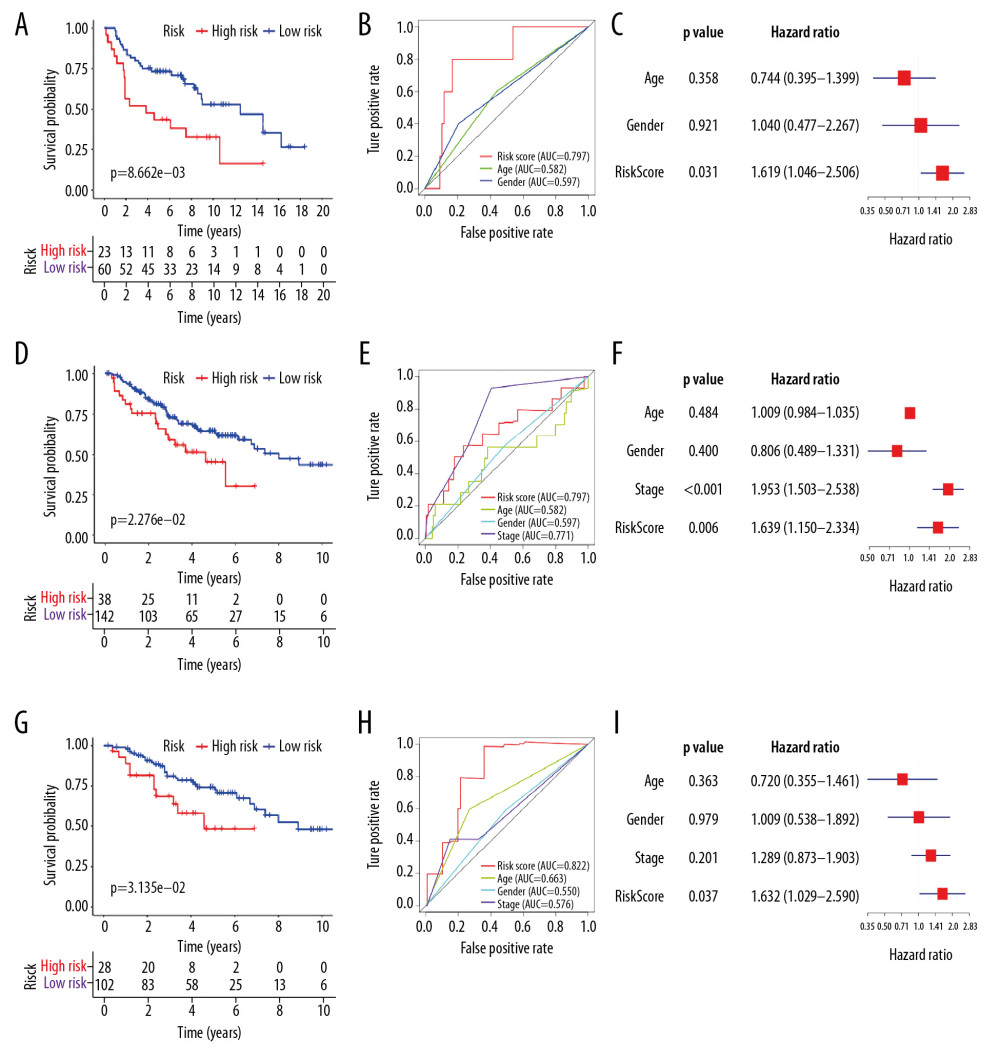 Figure 4. Predicted performance of the identified prognostic signature in the testing datasets (GSE30219: A–C; GSE41271: D–F; GSE42127: G–I). (A, D, G) The Kaplan-Meier survival analysis of the 7-gene prognostic signature. The same cutoff value in the training set is applied. (B, E, H) The multiple ROC curves comparing risk score with other clinical factors. ROC curve – receiver operating characteristic curve; AUC – area under the ROC curve. (C, F, I) The forest plots for multivariate survival analysis combining risk score and clinical factors. The hazard ratio and 95% confidence interval are presented.
Figure 4. Predicted performance of the identified prognostic signature in the testing datasets (GSE30219: A–C; GSE41271: D–F; GSE42127: G–I). (A, D, G) The Kaplan-Meier survival analysis of the 7-gene prognostic signature. The same cutoff value in the training set is applied. (B, E, H) The multiple ROC curves comparing risk score with other clinical factors. ROC curve – receiver operating characteristic curve; AUC – area under the ROC curve. (C, F, I) The forest plots for multivariate survival analysis combining risk score and clinical factors. The hazard ratio and 95% confidence interval are presented. 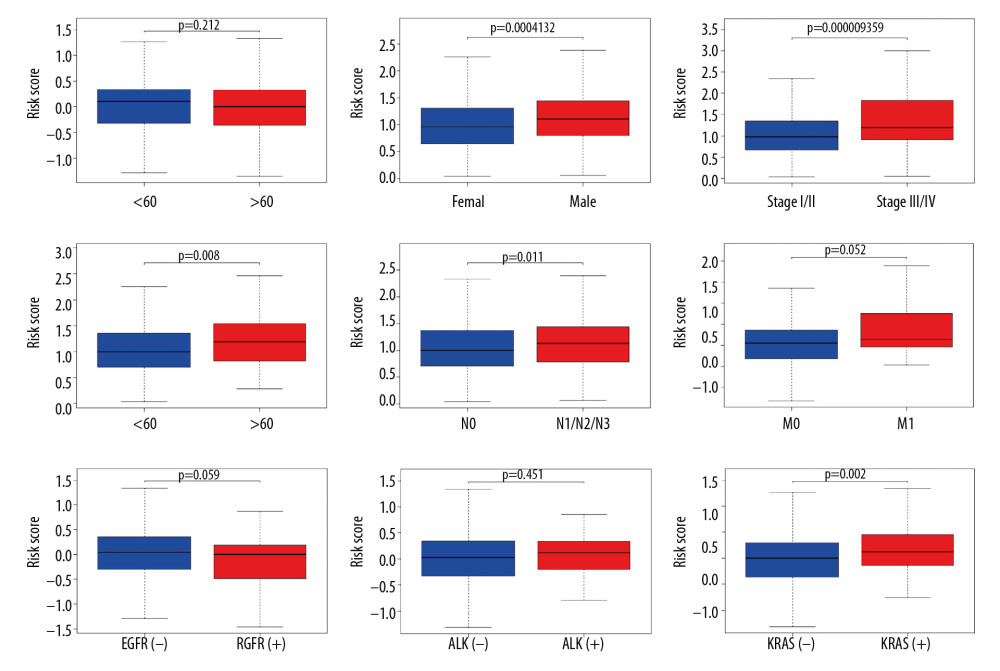 Figure 5. The boxplots for clinical correlation analysis of the prognostic signature in the TCGA-LUAD dataset. Factors including age, gender, TNM stage classification, and common gene mutations are taken into account for subgroup analyses. Horizontal line: mean value of risk score; vertical lines: the upper and lower quartiles. TCGA-LUAD – The Cancer Genome Atlas Lung Adenocarcinoma.
Figure 5. The boxplots for clinical correlation analysis of the prognostic signature in the TCGA-LUAD dataset. Factors including age, gender, TNM stage classification, and common gene mutations are taken into account for subgroup analyses. Horizontal line: mean value of risk score; vertical lines: the upper and lower quartiles. TCGA-LUAD – The Cancer Genome Atlas Lung Adenocarcinoma. 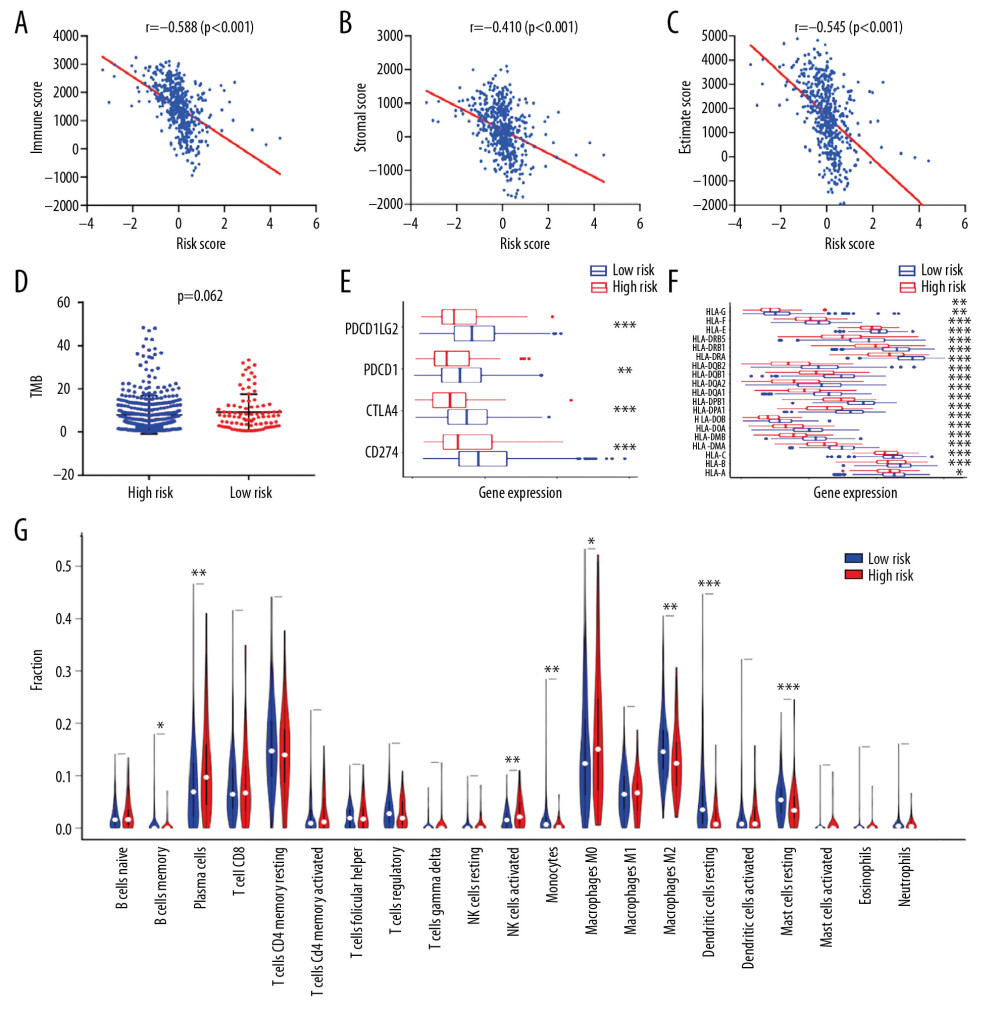 Figure 6. Immune-correlation analyses of the prognostic signature in the TCGA-LUAD dataset. (A–C) The correlation scatter plots representing the correlation between risk score and immune-, stromal, and ESTIMATE-score. The value of spearman correlation coefficient (r) is presented. (D) The comparison of the tumor mutation burden (TMB) values between high-risk (n=90) and low-risk (n=400) patients. (E) Gene expression levels of immune checkpoints. PDCD1LG2 – programmed cell death 1 ligand 2; PDCD1 – programmed cell death 1; CTLA4 – cytotoxic T-lymphocyte associated protein 4; CD274 – programmed cell death 1 ligand 1; * P-value <0.05. (F) Gene expression levels of human leukocyte antigen (HLA) system; * P-value <0.05. (G) The violin plot for the analysis of immune cell infiltration. The fractions of 22 mature human tumor infiltration leukocytes (TILs) are compared between the high-risk (n=70) and low-risk (n=372) groups. Naive CD4 T cells with negative infiltration in all samples is excluded from the analysis.
Figure 6. Immune-correlation analyses of the prognostic signature in the TCGA-LUAD dataset. (A–C) The correlation scatter plots representing the correlation between risk score and immune-, stromal, and ESTIMATE-score. The value of spearman correlation coefficient (r) is presented. (D) The comparison of the tumor mutation burden (TMB) values between high-risk (n=90) and low-risk (n=400) patients. (E) Gene expression levels of immune checkpoints. PDCD1LG2 – programmed cell death 1 ligand 2; PDCD1 – programmed cell death 1; CTLA4 – cytotoxic T-lymphocyte associated protein 4; CD274 – programmed cell death 1 ligand 1; * P-value <0.05. (F) Gene expression levels of human leukocyte antigen (HLA) system; * P-value <0.05. (G) The violin plot for the analysis of immune cell infiltration. The fractions of 22 mature human tumor infiltration leukocytes (TILs) are compared between the high-risk (n=70) and low-risk (n=372) groups. Naive CD4 T cells with negative infiltration in all samples is excluded from the analysis. 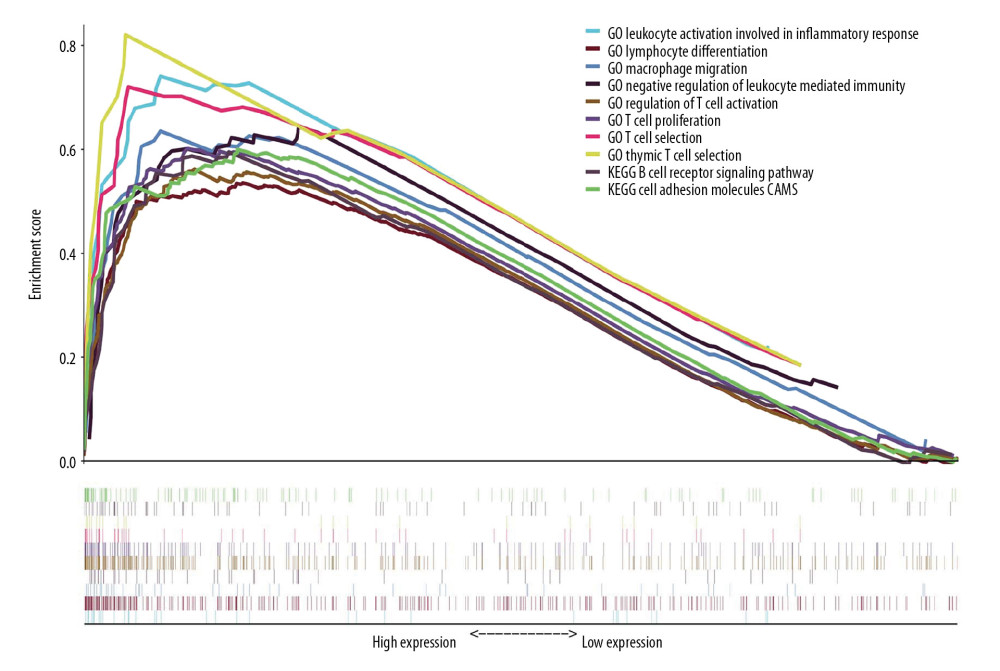 Figure 7. GSEA for the 7-gene signature comparing the high-risk and low-risk groups in the TCGA-LUAD dataset. The integrated enrichment plot lists 10 immune-associated pathways significantly enriched in the low-risk group (FDR <0.05) GSEA – gene set enrichment analysis; TCGA-LUAD – The Cancer Genome Atlas Lung Adenocarcinoma; FDR – false discovery rate; GO – Gene Ontology; KEGG – Kyoto Encyclopedia of Genes and Genomes.
Figure 7. GSEA for the 7-gene signature comparing the high-risk and low-risk groups in the TCGA-LUAD dataset. The integrated enrichment plot lists 10 immune-associated pathways significantly enriched in the low-risk group (FDR <0.05) GSEA – gene set enrichment analysis; TCGA-LUAD – The Cancer Genome Atlas Lung Adenocarcinoma; FDR – false discovery rate; GO – Gene Ontology; KEGG – Kyoto Encyclopedia of Genes and Genomes. References
1. Siegel RL, Miller KD, Jemal A, Cancer statistics, 2019: Cancer J Clin, 2019; 69(1); 7-34
2. Zappa C, Mousa SA, Non-small cell lung cancer: Current treatment and future advances: Transl Lung Cancer Res, 2016; 5(3); 288-300
3. Jao K, Tomasini P, Kamel-Reid S, The prognostic effect of single and multiple cancer-related somatic mutations in resected non-small-cell lung cancer: Lung Cancer, 2018; 123; 22-29
4. Testa U, Castelli G, Pelosi E, Lung cancers: Molecular characterization, clonal heterogeneity and evolution, and cancer stem cells: Cancers (Basel), 2018; 10(8) pii: E248
5. Lee SJ, Lee J, Park YS, Impact of smoking on mortality of patients with non-small cell lung cancer: Thorac Cancer, 2014; 5(1); 43-49
6. Slebos RJ, Kibbelaar RE, Dalesio O, K-ras oncogene activation as a prognostic marker in adenocarcinoma of the lung: N Engl J Med, 1990; 323(9); 561-65
7. Tas F, Ciftci R, Kilic L, Karabulut S, Age is a prognostic factor affecting survival in lung cancer patients: Oncol Lett, 2013; 6(5); 1507-13
8. Lim W, Ridge CA, Nicholson AG, Mirsadraee S: Quant Imaging Med Surg, 2018; 8(7); 709-18
9. Pezzuto A, Perrone G, Orlando N, A close relationship between HIF-1alpha expression and bone metastases in advanced NSCLC, a retrospective analysis: Oncotarget, 2019; 10(66); 7071-79
10. Pezzuto A, Cappuzzo F, D’Arcangelo M, Prognostic value of p16 protein in patients with surgically treated non-small cell lung cancer; relationship with Ki-67 and PD-L1: Anticancer Res, 2020; 40(2); 983-90
11. Ojlert AK, Halvorsen AR, Nebdal D, The immune microenvironment in non-small cell lung cancer is predictive of prognosis after surgery: Mol Oncol, 2019; 13(5); 1166-79
12. Givechian KB, Garner C, Benz S, An immunogenic NSCLC microenvironment is associated with favorable survival in lung adenocarcinoma: Oncotarget, 2019; 10(19); 1840-49
13. Pandey JP, Prognostic immune markers in non-small cell lung cancer – letter: Clin Cancer Res, 2011; 17(24); 7835 author reply 7836
14. Rossi G, Pezzuto A, Sini C, Concomitant medications during immune checkpoint blockage in cancer patients: Novel insights in this emerging clinical scenario: Crit Rev Oncol Hematol, 2019; 142; 26-34
15. Clough E, Barrett T, The gene expression omnibus database: Methods Mol Biol, 2016; 1418; 93-110
16. Chen B, Khodadoust MS, Liu CL, Profiling tumor infiltrating immune cells with CIBERSORT: Methods Mol Biol, 2018; 1711; 243-59
17. Yoshihara K, Shahmoradgoli M, Martinez E, Inferring tumour purity and stromal and immune cell admixture from expression data: Nat Commun, 2013; 4; 2612
18. Ritchie ME, Phipson B, Wu D, Limma powers differential expression analyses for RNA-sequencing and microarray studies: Nucleic Acids Res, 2015; 43(7); e47
19. Friedman J, Hastie T, Tibshirani R, Regularization paths for generalized linear models via coordinate descent: J Stat Softw, 2010; 33(1); 1-22
20. Simon N, Friedman J, Hastie T, Tibshirani R, Regularization paths for Cox’s proportional hazards model via coordinate descent: J Stat Softw, 2011; 39(5); 1-13
21. Camp RL, Dolled-Filhart M, Rimm DL, X-tile: A new bio-informatics tool for biomarker assessment and outcome-based cut-point optimization: Clin Cancer Res, 2004; 10(21); 7252-59
22. Allgauer M, Budczies J, Christopoulos P, Implementing tumor mutational burden (TMB) analysis in routine diagnostics – a primer for molecular pathologists and clinicians: Transl Lung Cancer Res, 2018; 7(6); 703-15
23. Pienta KJ, McGregor N, Axelrod R, Axelrod DE, Ecological therapy for cancer: Defining tumors using an ecosystem paradigm suggests new opportunities for novel cancer treatments: Transl Oncol, 2008; 1(4); 158-64
24. Swartz MA, Iida N, Roberts EW, Tumor microenvironment complexity: Emerging roles in cancer therapy: Cancer Res, 2012; 72(10); 2473-80
25. Fridman WH, Pages F, Sautes-Fridman C, Galon J, The immune contexture in human tumours: impact on clinical outcome: Nat Rev Cancer, 2012; 12(4); 298-306
26. Fridman WH, Zitvogel L, Sautes-Fridman C, Kroemer G, The immune contexture in cancer prognosis and treatment: Nat Rev Clin Oncol, 2017; 14(12); 717-34
27. Remark R, Becker C, Gomez JE, The non-small cell lung cancer immune contexture. A major determinant of tumor characteristics and patient outcome: Am J Respir Crit Care Med, 2015; 191(4); 377-90
28. Broussard EK, Disis ML, TNM staging in colorectal cancer: T is for T cell and M is for memory: J Clin Oncol, 2011; 29(6); 601-3
29. Takanami I, Takeuchi K, Kodaira S, Tumor-associated macrophage infiltration in pulmonary adenocarcinoma: Association with angiogenesis and poor prognosis: Oncology (Basel), 1999; 57(2); 138-42
30. Villegas FR, Coca S, Villarrubia VG, Prognostic significance of tumor infiltrating natural killer cells subset CD57 in patients with squamous cell lung cancer: Lung Cancer, 2002; 35(1); 23-28
31. Kikuchi E, Yamazaki K, Torigoe T, HLA class I antigen expression is associated with a favorable prognosis in early stage non-small cell lung cancer: Cancer Sci, 2007; 98(9); 1424-30
32. Mu CY, Huang JA, Chen Y, High expression of PD-L1 in lung cancer may contribute to poor prognosis and tumor cells immune escape through suppressing tumor infiltrating dendritic cells maturation: Med Oncol, 2011; 28(3); 682-88
33. Nixon AB, Schalper KA, Jacobs I, Peripheral immune-based biomarkers in cancer immunotherapy: Can we realize their predictive potential?: J Immunother Cancer, 2019; 7(1); 325
34. Nevins JR, Potti A, Mining gene expression profiles: Expression signatures as cancer phenotypes: Nat Rev Genet, 2007; 8(8); 601-9
35. Sini C, Tuzi A, Rossi G, Acquired resistance in oncogene-addicted non-small-cell lung cancer: Future Oncol, 2018; 14(13s); 29-40
36. Dong ZY, Zhong WZ, Zhang XC, Potential predictive value of TP53 and KRAS mutation status for response to PD-1 blockade immunotherapy in lung adenocarcinoma: Clin Cancer Res, 2017; 23(12); 3012-24
37. Reith W, LeibundGut-Landmann S, Waldburger JM, Regulation of MHC class II gene expression by the class II transactivator: Nat Rev Immunol, 2005; 5(10); 793-806
38. Thibodeau J, Bourgeois-Daigneault MC, Lapointe R, Targeting the MHC class II antigen presentation pathway in cancer immunotherapy: Oncoimmunology, 2012; 1(6); 908-16
39. van den Elsen PJ, van der Stoep N, Class II transactivator (CIITA) deficiency in tumor cells: complicated mechanisms or not?: Am J Pathol, 2003; 163(1); 373-75 author reply 375–76
40. He Y, Rozeboom L, Rivard CJ, MHC class II expression in lung cancer: Lung Cancer, 2017; 112; 75-80
41. Dambuza IM, Brown GD, C-type lectins in immunity: recent developments: Curr Opin Immunol, 2015; 32; 21-27
42. Chiba S, Ikushima H, Ueki H, Recognition of tumor cells by Dectin-1 orchestrates innate immune cells for anti-tumor responses: Elife, 2014; 3; e04177
43. Kutikhin AG, Yuzhalin AE, C-type lectin receptors and RIG-I-like receptors: New points on the oncogenomics map: Cancer Manag Res, 2012; 4; 39-53
44. Lev S, Zeevi DA, Frumkin A, Constitutive activity of the human TRPML2 channel induces cell degeneration: J Biol Chem, 2010; 285(4); 2771-82
45. Rinkenberger N, Schoggins JW, Mucolipin-2 cation channel increases trafficking efficiency of endocytosed viruses: mBio, 2018; 9(1) pii: e02314-17
46. Valadez JA, Cuajungco MP, PAX5 is the transcriptional activator of mucolipin-2 (MCOLN2) gene: Gene, 2015; 555(2); 194-202
47. Li YL, Zhao H, Ren XB, Relationship of VEGF/VEGFR with immune and cancer cells: staggering or forward?: Cancer Biol Med, 2016; 13(2); 206-14
48. Carrillo de Santa Pau E, Arias FC, Caso Pelaez E, Prognostic significance of the expression of vascular endothelial growth factors A, B, C, and D and their receptors R1, R2, and R3 in patients with non-small cell lung cancer: Cancer, 2009; 115(8); 1701-12
49. Rossi D, Deaglio S, Dominguez-Sola D, Alteration of BIRC3 and multiple other NF-kappaB pathway genes in splenic marginal zone lymphoma: Blood, 2011; 118(18); 4930-34
50. Conte D, Holcik M, Lefebvre CA, Inhibitor of apoptosis protein cIAP2 is essential for lipopolysaccharide-induced macrophage survival: Mol Cell Biol, 2006; 26(2); 699-708
51. Bertrand MJ, Doiron K, Labbe K, Cellular inhibitors of apoptosis cIAP1 and cIAP2 are required for innate immunity signaling by the pattern recognition receptors NOD1 and NOD2: Immunity, 2009; 30(6); 789-801
52. D’Andrea EL, Ferravante A, Scudiero I, The dishevelled, EGL-10 and pleckstrin (DEP) domain-containing protein DEPDC7 binds to CARMA2 and CARMA3 proteins, and regulates NF-kappaB activation: PLoS One, 2014; 9(12); e116062
53. Lee JH, Choi SI, Kim RK, Tescalcin/c-Src/IGF1Rbeta-mediated STAT3 activation enhances cancer stemness and radioresistant properties through ALDH1: Sci Rep, 2018; 8(1); 10711
Figures
Figure 1. The flow chart of study design.Figure 2. Predicted performance of the identified prognostic signature in the training dataset. (A) The Kaplan-Meier survival analysis of the 7-gene prognostic signature in the TCGA-LUAD cohort. According to the optimal cutoff value, samples are divided into high-risk and low-risk groups. (B) The calibration curves of the model for predicting CSS in LUAD. The predicted 3-year survival probability is plotted on the X-axis; the corresponding actual survival rate is plotted on the Y-axis. (C) The distribution plot of risk score. Patients are arranged from left to right in an increasing order of risk score. (D) The survival status of each patient. Y-axis represents the overall survival time. The color code: green for alive case, red for dead case. (E) The heatmap of mRNA expression levels of the 7 genes. CSS – cancer specific survival; LUAD – lung adenocarcinoma.Figure 3. (A) The ROC curves comparing the prognostic values of risk score and several clinical factors in the TCGA-LUAD dataset. The predicted time point is 1 year. ROC curve – receiver operating characteristic curve; AUC – area under the ROC curve; TCGA-LUAD – The Cancer Genome Atlas Lung Adenocarcinoma. (B) The forest plot for multivariate survival analysis based on risk score and other clinical factors in TCGA-LUAD. The hazard ratio and 95% confidence interval are presented. (C) The nomogram constructed for the prediction of survival probability.Figure 4. Predicted performance of the identified prognostic signature in the testing datasets (GSE30219: A–C; GSE41271: D–F; GSE42127: G–I). (A, D, G) The Kaplan-Meier survival analysis of the 7-gene prognostic signature. The same cutoff value in the training set is applied. (B, E, H) The multiple ROC curves comparing risk score with other clinical factors. ROC curve – receiver operating characteristic curve; AUC – area under the ROC curve. (C, F, I) The forest plots for multivariate survival analysis combining risk score and clinical factors. The hazard ratio and 95% confidence interval are presented.Figure 5. The boxplots for clinical correlation analysis of the prognostic signature in the TCGA-LUAD dataset. Factors including age, gender, TNM stage classification, and common gene mutations are taken into account for subgroup analyses. Horizontal line: mean value of risk score; vertical lines: the upper and lower quartiles. TCGA-LUAD – The Cancer Genome Atlas Lung Adenocarcinoma.Figure 6. Immune-correlation analyses of the prognostic signature in the TCGA-LUAD dataset. (A–C) The correlation scatter plots representing the correlation between risk score and immune-, stromal, and ESTIMATE-score. The value of spearman correlation coefficient (r) is presented. (D) The comparison of the tumor mutation burden (TMB) values between high-risk (n=90) and low-risk (n=400) patients. (E) Gene expression levels of immune checkpoints. PDCD1LG2 – programmed cell death 1 ligand 2; PDCD1 – programmed cell death 1; CTLA4 – cytotoxic T-lymphocyte associated protein 4; CD274 – programmed cell death 1 ligand 1; * P-value <0.05. (F) Gene expression levels of human leukocyte antigen (HLA) system; * P-value <0.05. (G) The violin plot for the analysis of immune cell infiltration. The fractions of 22 mature human tumor infiltration leukocytes (TILs) are compared between the high-risk (n=70) and low-risk (n=372) groups. Naive CD4 T cells with negative infiltration in all samples is excluded from the analysis.Figure 7. GSEA for the 7-gene signature comparing the high-risk and low-risk groups in the TCGA-LUAD dataset. The integrated enrichment plot lists 10 immune-associated pathways significantly enriched in the low-risk group (FDR <0.05) GSEA – gene set enrichment analysis; TCGA-LUAD – The Cancer Genome Atlas Lung Adenocarcinoma; FDR – false discovery rate; GO – Gene Ontology; KEGG – Kyoto Encyclopedia of Genes and Genomes. Tables
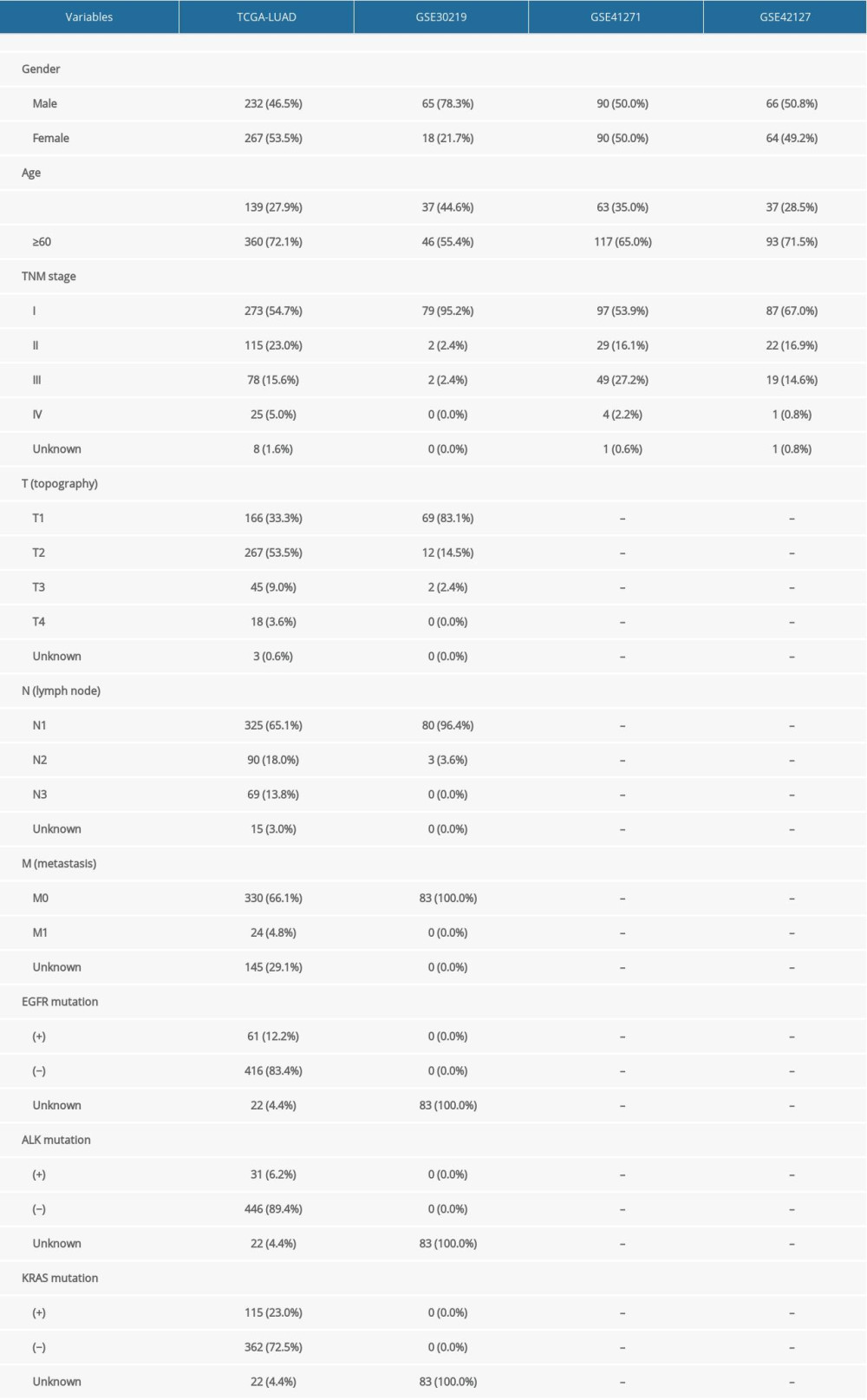 Table 1. Demographic and clinical characteristics of subjects in the four datasets.
Table 1. Demographic and clinical characteristics of subjects in the four datasets.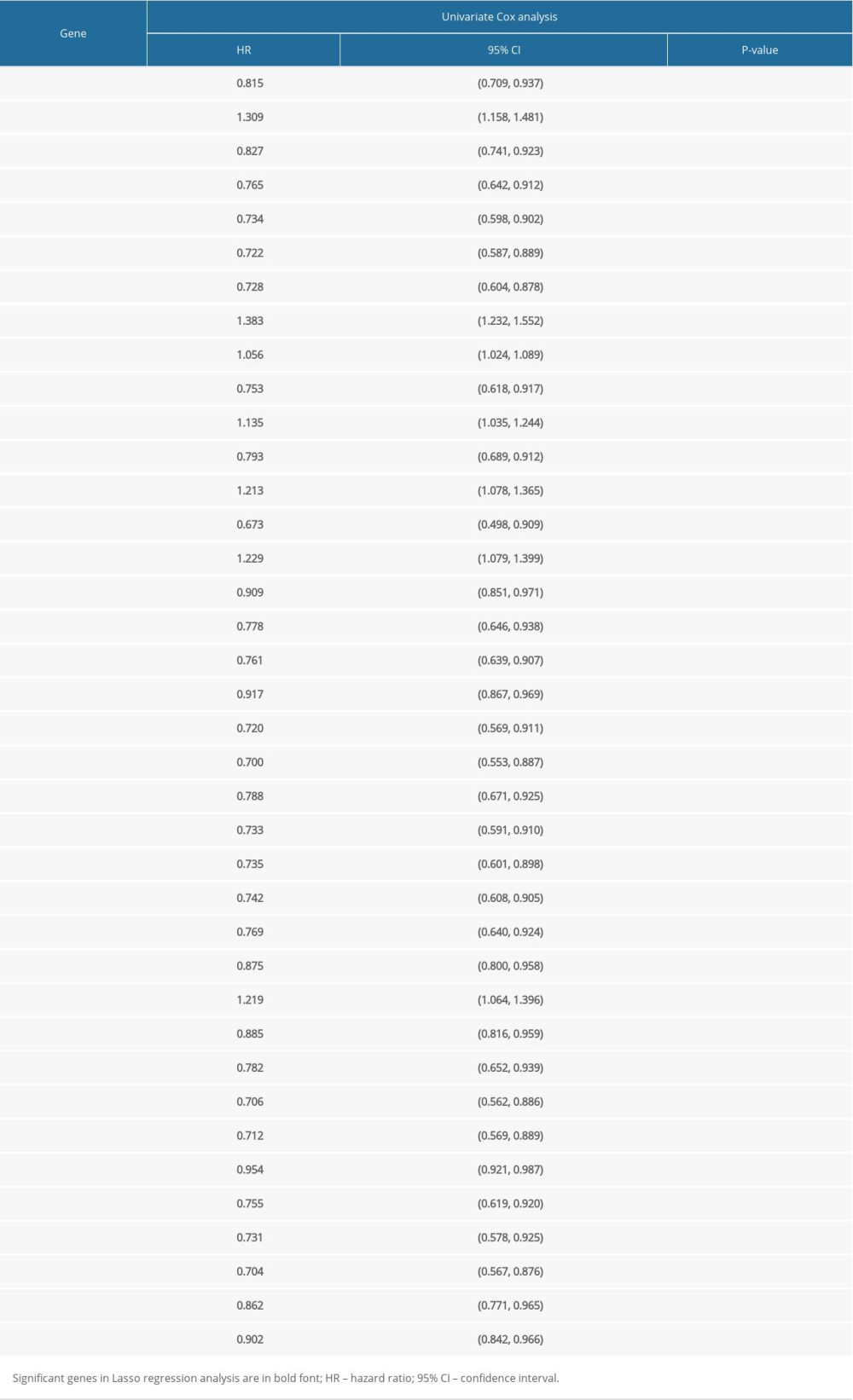 Table 2. Univariate Cox regression analysis of the 38 identified genes.
Table 2. Univariate Cox regression analysis of the 38 identified genes.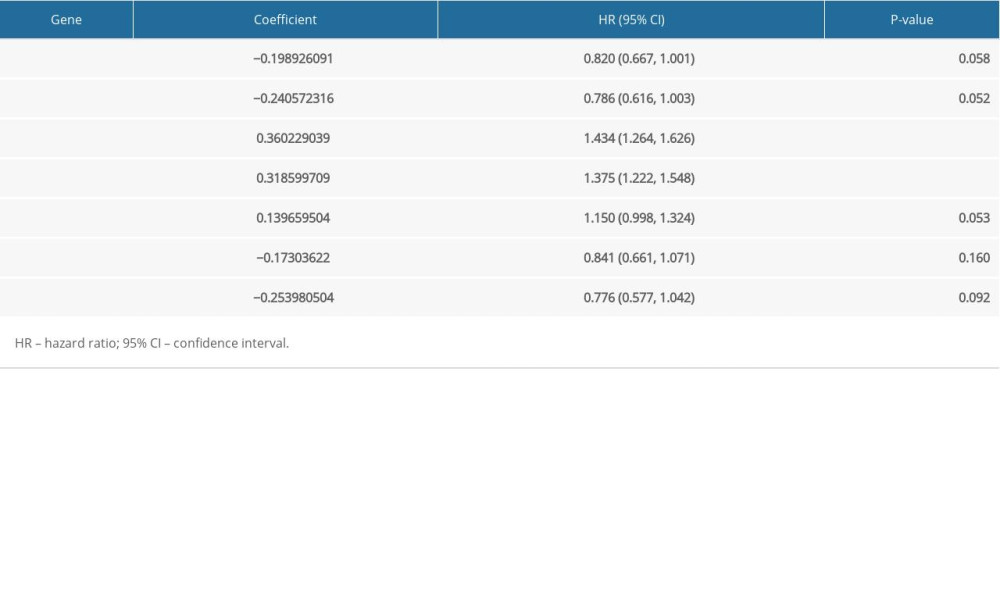 Table 3. Multi-variate Cox regression analysis of the 7-gene signature.Table 1. Demographic and clinical characteristics of subjects in the four datasets.Table 2. Univariate Cox regression analysis of the 38 identified genes.Table 3. Multi-variate Cox regression analysis of the 7-gene signature.
Table 3. Multi-variate Cox regression analysis of the 7-gene signature.Table 1. Demographic and clinical characteristics of subjects in the four datasets.Table 2. Univariate Cox regression analysis of the 38 identified genes.Table 3. Multi-variate Cox regression analysis of the 7-gene signature. In Press
Clinical Research
Effects of Single-Bout Endurance Exercise Intensity on Peripheral Neurotrophic Factors in Patients With Isc...Med Sci Monit In Press; DOI: 10.12659/MSM.952089
Review article
Anisodus tanguticus in Cancer Research: A Review of Traditional Use, Phytochemistry, Extraction Methods, an...Med Sci Monit In Press; DOI: 10.12659/MSM.952999
Clinical Research
Nasal Mucociliary Clearance and Its Relationship With Disease Severity in Patients With Multiple SclerosisMed Sci Monit In Press; DOI: 10.12659/MSM.952850
Clinical Research
Modified Thoracoabdominal Nerves Block Through the Perichondrial Approach vs Subcostal Transversus Abdomini...Med Sci Monit In Press; DOI: 10.12659/MSM.953976
Most Viewed Current Articles
17 Jan 2024 : Review article 14,176,570
Vaccination Guidelines for Pregnant Women: Addressing COVID-19 and the Omicron VariantDOI :10.12659/MSM.942799
Med Sci Monit 2024; 30:e942799
13 Nov 2021 : Clinical Research 3,762,188
Acceptance of COVID-19 Vaccination and Its Associated Factors Among Cancer Patients Attending the Oncology ...DOI :10.12659/MSM.932788
Med Sci Monit 2021; 27:e932788
14 Dec 2022 : Clinical Research 2,466,310
Prevalence and Variability of Allergen-Specific Immunoglobulin E in Patients with Elevated Tryptase LevelsDOI :10.12659/MSM.937990
Med Sci Monit 2022; 28:e937990
16 May 2023 : Clinical Research 708,927
Electrophysiological Testing for an Auditory Processing Disorder and Reading Performance in 54 School Stude...DOI :10.12659/MSM.940387
Med Sci Monit 2023; 29:e940387