31 August 2020: Database Analysis
, , and are Prognostic Biomarkers and Correlated with Immune Infiltration in Hepatocellular Carcinoma
Yiping Zou12CE, Shiye Ruan1CD, Liang Jin1C, Zhihong Chen1BF, Hongwei Han1DF, Yuanpeng Zhang1F, Zhixiang Jian1G, Ye Lin1D, Ning Shi1AE, Haosheng Jin1AG*DOI: 10.12659/MSM.925289
Med Sci Monit 2020; 26:e925289






Abstract
BACKGROUND: Orderly G2/M transition in the cell cycle is controlled by the cyclin-dependent kinase 1/cyclin B (CDK1/CCNB) complex. We aimed to comprehensively investigate the roles of CDK1, CCNB1, and CCNB2 via multi-omics analysis and their relationships with immune infiltration in hepatocellular carcinoma (HCC).
MATERIAL AND METHODS: The transcriptional data and the epigenetic and genetic alterations of CDK1, CCNB1, and CCNB2, as well as their impacts on prognosis in HCC patients, were identified using multiple databases. The correlations between expression of these genes and immune infiltration in HCC were then explored using the TIMER database.
RESULTS: Overall, mRNA expression of CDK1, CCNB1, and CCNB2 was up-regulated in various tumor tissues including HCC. Higher expression of these genes was associated with poorer prognosis in HCC patients. Lower promoter methylation of these genes might cause higher expression levels in tumor tissues of HCC. Genetic alterations and several methylated-CpG sites in these genes were significantly associated with survival. Notably, expression levels of CDK1, CCNB1, and CCNB2 were positively correlated with infiltrating levels of CD4⁺ T cells, CD8⁺ T cells, neutrophils, macrophages, and dendritic cells in HCC. In addition, significant correlations between the expression of these genes and various immune markers in HCC, such as PD-1, PDL-1, and CTLA-4, were also observed.
CONCLUSIONS: CDK1, CCNB1, and CCNB2 are potential prognostic biomarkers and associated with immune cell infiltration in HCC. The genes may be utilized to predict the reaction of immunotherapy. Combining inhibitors of these genes with immunotherapy may improve the survival time of HCC patients.
Keywords: Biological Markers, Carcinoma, Hepatocellular, CDC2 Protein Kinase, Tumor Escape, Biomarkers, Tumor, Cyclin B1, Cyclin B2, Liver Neoplasms
Background
Liver cancer is one of the most common types of cancer and the third leading cause of cancer death worldwide. Hepatocellular carcinoma (HCC), which composes 75% to 85% of primary liver cancer cases, is the major pathological type [1]. HCC is often secondary to chronic liver cirrhosis, and various risk factors have been identified, such as chronic hepatitis B virus (HBV) infection, hepatitis C virus (HCV) infection, autoimmune hepatitis, alcohol abuse, and several metabolic diseases [2,3]. Current therapeutic strategies for HCC, such as surgical resection, liver transplantation, and radiofrequency ablation, have been widely used and improved, but the long-term survival rate of HCC is still unsatisfactory due to the high percentage of cases that are at an advanced stage at diagnosis and the high recurrence rate after surgical resection [4,5]. Only 2 first-line therapeutic drugs, the oral multikinase inhibitors sorafenib and lenvatinib, have shown clinical benefits in patients with advanced HCC [6,7]. Therefore, in order to improve survival of HCC patients, it is critical to evaluate carcinogenesis mechanisms and explore potential drug targets.
Cyclin B1 (CCNB1) and cyclin B2 (CCNB2) can form complexes with CDK1 (cyclin-dependent kinase 1) to regulate the G2/M phases of the mammalian cell cycle, which plays an important role in the initiation of mitosis [8]. Dysregulation of CDKs is associated with the uncontrolled cell proliferation in human cancers. In addition, higher expression of cyclin B is associated with poorer outcomes for gastric, esophageal, breast, and non-small-cell lung cancer [9]. As shown in recent research, the knockdown of
In the current study, we used several public databases to comprehensively analyze the expression levels of
Material and methods
THE DESCRIPTION OF ALL DATABASES:
All bioinformatics analyses were based on sequencing or microarray data obtained from tumor tissues and corresponding normal tissues. The data in this study involved samples from The Cancer Genome Atlas (TCGA), the International Cancer Genome Consortium (ICGC), and the Gene Expression Omnibus (GEO). Figure 1E shows the number of samples from different datasets. Samples with missing data on survival information and clinical information such as tumor pathological grade were excluded from survival analysis and corresponding subgroup analyses. Using various online database analysis tools, we explored the transcription of CDK1, CCNB1, and CCNB2 in various tumors and the prognostic values of these genes in HCC. In addition, we identified the promoter methylation and genetic alterations of CDK1, CCNB1, and CCNB2 and their impacts of prognosis in HCC patients. Finally, we explored the correlations between expression of these genes and immune infiltration in HCC. The specific online databases and the statistical methods used are as follows.
ONCOMINE DATABASE ANALYSIS:
ONCOMINE (https://www.oncomine.org/resource/main.html) is an online tumor data analysis platform [17]. By using the Oncomine database 4.5, we identified the mRNA expression levels of the CDK1, CCNB1, and CCNB2 genes in various cancers.
HCCDB DATABASE ANALYSIS:
HCCDB (http://lifeome.net/database/hccdb/home.html) is a novel database containing 3917 samples, which were obtained from 15 public datasets of gene expression in HCC [18]. The database includes 2 RNA-Seq datasets (TCGA and ICGC) and 13 microarray datasets from GEO. In our study, HCCDB was used to provide the visualization for differential expression analysis of CDK1, CCNB1, and CCNB2 in HCC from several datasets.
UALCAN DATABASE ANALYSIS:
UALCAN (http://ualcan.path.uab.edu) is a publicly available interactive online portal that is used to analyze the relative expression and methylation of genes in normal and tumor tissues from TCGA [19]. Subgroups based on pathological grades and individual cancer stages can be further investigated in depth. Histological grades of HCC were defined as follows: well-differentiated (I), moderately differentiated (II), poorly differentiated (III), and undifferentiated (IV). Individual cancer stages were based on the American Joint Committee on Cancer (AJCC) stage.
GEPIA DATASET ANALYSIS:
GEPIA (http://gepia.cancer-pku.cn/) is an online database that contains expression data based on 9736 tumors and 8587 normal tissues from GTEx and TCGA [20]. We used this dataset to analyze the correlations between expression of CDK1, CCNB1, and CCNB2 and survival, including overall survival (OS) and disease-free survival (DFS). Furthermore, gene expression correlation analyses of those 3 genes were performed based on the TCGA expression dataset.
LINKEDOMICS DATABASE ANALYSIS:
The LinkedOmics database (http://www.linkedomics.org/login.php) is an easy-to-use online tool for analyzing 32 TCGA cancer-associated multidimensional datasets [21]. Using this database, we analyzed genes that were positively correlated with CDK1, CCNB1, and CCNB2. Heat maps were applied to show the top 50 positively correlated genes.
GO ANALYSIS AND KEGG ANALYSIS:
Based on the co-occurrence genes from the LinkedOmics database, Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses were performed, using the online tool DAVID (
METHSURV DATABASE ANALYSIS:
MethSurv (https://biit.cs.ut.ee/methsurv) is a visualization web tool used to investigate methylation biomarkers associated with survival in different types of cancer [22]. By analyzing the TCGA methylation data from the MethSurv, we obtained the prognostic value of each DNA methylation CpG site for CDK1, CCNB1, and CCNB2 in HCC.
C-BIOPORTAL DATABASE ANALYSIS:
The c-BioPortal (https://www.cbioportal.org) is an online database for analyzing multidimensional cancer genomics data [23]. We used the dataset that included 360 cases of liver hepatocellular carcinoma (LIHC) from TCGA. Using the c-BioPortal, we identified mutations, copy-number alterations (CNAs), and mRNA expression of CDK1, CCNB1, and CCNB2 in HCC. Furthermore, we assessed the relationships between the degree of methylation of the 3 genes and their mRNA expression levels.
TIMER DATABASE ANALYSIS:
TIMER (https://cistrome.shinyapps.io/timer/) is a useful web server for analyzing the immune infiltration in different types of cancer from the TCGA database [24]. By applying a deconvolution method, TIMER determines the infiltration levels of immune cells from gene expression profiles in tumor tissues. We analyzed the correlation of CDK1, CCNB1, and CCNB2 expression in HCC with the infiltrating levels of immune cells, respectively, including CD4+ T cells, CD8+ T cells, B cells, macrophages, dendritic cells (DCs), and neutrophils. In addition, we investigated the genetic markers for immune cell infiltration in tumors, including tumor-associated macrophages (TAMs), M1 macrophages, M2 macrophages, DCs, neutrophils, T-helper 1 (Th1) cells, T-helper (Th2) cells, regulatory T cells (Tregs), natural killer (NK) cells, and B cell and T cell exhaustion.
STATISTICAL ANALYSIS:
Expression levels of
Results
:
Using the ONCOMINE database, we analyzed the mRNA expression levels for CDK1, CCNB1, and CCNB2 in different types of cancer. The results showed that compared with normal tissues, the levels were significantly higher in several types of tumor tissues, such as liver cancer, kidney cancer, breast cancer, colorectal cancer, gastric cancer, esophageal cancer, and lung cancer, among others (Figure 1A). We further used the TIMER database to analyze the RNA-seq data from the TCGA database. The result also confirmed that mRNA expression levels of CDK1, CCNB1, and CCNB2 were significantly higher in LIHC, kidney renal clear cell carcinoma, esophageal carcinoma, colon adenocarcinoma, lung adenocarcinoma, stomach adenocarcinoma, and so forth than in the adjacent normal tissues (Figure 1B–1D). Using the HCCDB databases, we evaluated the expression levels reported in HCC studies from GEO, TCGA, and ICGC. Among these studies, 11 datasets indicated that mRNA expression levels of CDK1 and CCNB2 were significantly higher in the HCC tissues than in the adjacent normal tissues. In addition, 12 datasets revealed that mRNA expression levels of CCNB1 in the HCC tissues were significantly higher than in the adjacent normal tissues (Figure 1E–1G).
:
We next explored the different expression levels of the 3 genes in HCC, stratified according to the AJCC stage and pathological grade. The results demonstrated that the expression of the 3 genes was higher in HCC tissues than in normal tissues based on different pathological grades and individual cancer stages. Therefore, expression levels of CDK1, CCNB1, and CCNB2 may serve as potential diagnostic markers in patients with HCC. Furthermore, overexpression of these genes was also related to advanced pathological grades and individual cancer stages, except stage IV (with only 6 cases) (Figure 2A–2F). Therefore, the results indicated that expression of these genes plays an important role in the tumorigenesis and progression of HCC.
:
Using the data for LIHC from TCGA in the GEPIA database, we assessed the correlation between differential expression of CDK1, CCNB1, and CCNB2 and clinical outcomes. Based on results from 364 HCC patients, poorer prognosis in terms of DFS and OS (P<0.05), except OS of CCNB2 (P=0.052), were associated with higher mRNA expression levels for CDK1, CCNB1, and CCNB2 (Figure 3A–3F). We further investigated the prognostic value of these genes in the ICGC dataset by using the HCCDB database. The results further confirmed that increased expression of CDK1, CCNB1, and CCNB2 was significantly associated with poor OS in HCC (P<0.05) (Figure 3G–3I). Hence, higher expression levels of CDK1, CCNB1, and CCNB2 are indicators of poor survival for patients with HCC.
ANALYSIS OF CO-EXPRESSED GENES IN HCC:
We next analyzed the correlation between expression levels of CDK1, CCNB1, and CCNB2 in HCC using the GEPIA database. The results indicated that these 3 genes were significantly positively correlated: CDK1 and CCNB1 (r=0.91, P<0.001), CDK1 and CCNB2 (r=0.92, P<0.001), and CCNB1 and CCNB2 (r=0.93, P<0.001) (Figure 4A–4C). Genes positively co-expressed with CDK1, CCNB1, and CCNB2 were analyzed in the LIHC cohort from TCGA by using the LinkedOmics database. The top 50 genes that were significantly correlated with these 3 genes are shown in the heat maps (Figure 4D–4F). This result further confirmed that CDK1, CCNB1, and CCNB2 were strongly positively co-expressed in HCC. We combined genes that were co-expressed with these 3 genes, deleted duplicate values, and identified 62 co-expressed genes, which were then extracted for GO and KEGG analysis. We showed the top 5 GO of BPs, CCs, and MFs based on the minimum values of false discovery rate (FDR) and maximum counts of GO. The results indicated that the most significant BPs, CCs, and MFs were cell cycle, non-membrane-bound organelles, and ATP binding, respectively (Figure 4G). KEGG analysis revealed the key pathways of these genes: cell cycle, oocyte meiosis, progesterone-mediated oocyte maturation, and the p53 signaling pathway (Figure 4H).
:
Moreover, by analyzing the LIHC samples from TCGA in the UALCAN database, we evaluated the levels of CDK1, CCNB1, and CCNB2 promoter methylation in HCC and normal tissues. The results suggested that the levels of methylation were lower in HCC than in normal tissues (P<0.05) (Figure 5A–5C). In addition, we assessed the relationships between the degree of methylation of these 3 genes and the mRNA levels using the c-BioPortal database. The results indicated significant negative correlations between the methylation levels and the mRNA expression levels of these genes in HCC (P<0.05) (Figure 5D–5F). Thus, the results suggest that lower levels of CDK1, CCNB1, and CCNB2 promoter methylation might cause higher expression levels of these genes in HCC. In addition, the prognostic values associated with diverse CpG sites were also analyzed via the MethSurv database. Ultimately, the results showed that 7 CpG sites in CDK1, 6 CpG sites in CCNB1, and 2 CpG sites in CCNB2 were significantly associated with the prognosis of patients with HCC (Table 1).
:
The frequency and types of genetic alterations in CDK1, CCNB1, and CCNB2 in patients with HCC were analyzed by using the cBioPortal database. A total of 360 LIHC cases from TCGA were explored. CDK1, CCNB1, and CCNB2 were altered in 6%, 9%, and 9% of LIHC cases, respectively. The most frequent alteration type in these samples was mRNA upregulation (10.8%) (Figure 6A). Furthermore, the Kaplan-Meier curves for the altered and unaltered groups of these genes demonstrated significant differences in OS (P<0.001) and DFS (P=0.0368) in patients with HCC (Figure 6B, 6C). Therefore, genomic alterations of these genes were considered as poor prognosis factors in HCC patients.
:
By using the TIMER database, we explored whether the mRNA expression levels of CDK1, CCNB1, and CCNB2 were correlated with infiltrating immune cells in HCC. The results demonstrated that overexpression of each of these genes was significantly associated with higher immune cell infiltration levels. Specifically, the CDK1 expression level was positively correlated with infiltration levels of CD8+ T cells (r=0.316, P=2.38e–09), CD4+ T cells (r=0.332, P=2.72e–10), B cells (r=0.469, P=2.97e–20), macrophages (r=0.449, P=2.60e–18), neutrophils (r=0.344, P=4.98e–11), and DCs (r=0.442, P =1.17e–17) (Figure 7A). The CCNB1 expression level was positively correlated with infiltration levels of CD8+ T cells (r=0.303, P=1.12e–08), CD4+ T cells (r=0.283, P=9.33e–08), B cells (r=0.469, P =2.9e–20), macrophages (r=0.42, P=5.42e–16), neutrophils (r=0.342, P=6.81e–11), and DCs (r=0.429, P =1.15e–16) (Figure 7B). Similarly, the CCNB2 expression level was also positively correlated with infiltration levels of CD8+ T cells (r=0.35, P=2.78e–11), CD4+ T cells (r=0.305, P=7.86e–09), B cells (r=0.487, P=6.57e–22), macrophages (r=0.444, P=6.27e–18), neutrophils (r=0.318, P=1.49e–09), and DCs (r=0.457, P=6.57e–19) (Figure 7C). The results provided strong evidence that these genes play crucial roles for various immune infiltration cells, including CD4+ T cells, CD8+ T cells, B cells, neutrophils, macrophages, and DCs.
Moreover, the relationships between somatic copy number alterations (SCNA) of the 3 genes and tumor infiltration levels among HCC were investigated. Interestingly, the results showed that the CNA of CDK1 had significant correlations with the infiltration levels of CD4+ T cells and B cells; the CNA of CCNB1 had significant correlations with CD4+ T cells, neutrophils, and macrophages; and the CNA of CCNB2 had a significant correlation with CD4+ T cells (Figure 7D).
:
To further explore the relationships between CDK1, CCNB1, and CCNB2 expression and various immune infiltrating cells, we explored the correlations between these genes and immune markers for different subsets of immune cells in HCC. The immune markers analyzed in our study were used to characterize immune cells, including TAMs, monocytes, M1 and M2 macrophages, neutrophils, DCs, NK cells, and B cells. We also investigated diverse functional T cells, such as Th1, Th2, Tregs, and exhausted T cells. After the correlation was adjusted by tumor purity, the expression levels of CDK1, CCNB1, and CCNB2 were found to be significantly correlated with diverse immune markers in different immune cells in HCC (Table 2).
Notably, the expression levels of most immune markers for TAMs, monocytes, and M2 macrophages had moderate correlations with
Interestingly, the correlations between markers for DCs and the expression levels of
Discussion
Orderly G2/M transition is controlled by the CDK1/CCNB complex, which plays a critical role in governing the cell cycle of mammalian cells [8]. Recent studies reported that the mRNA expression levels of
Our study further proved overexpression of
Interestingly, another important finding from our study was that the mRNA expression levels of
Clinical trials of immune checkpoint inhibitors in HCC, such as tremelimumab (anti-CTLA4), nivolumab (anti-PD1), and durvalumab (anti-PDL1), have shown positive responses [31]. Our results indicate a poorer prognosis with higher expression levels of
Conclusions
Our study provides multi-pronged evidence of the impacts of
Figures
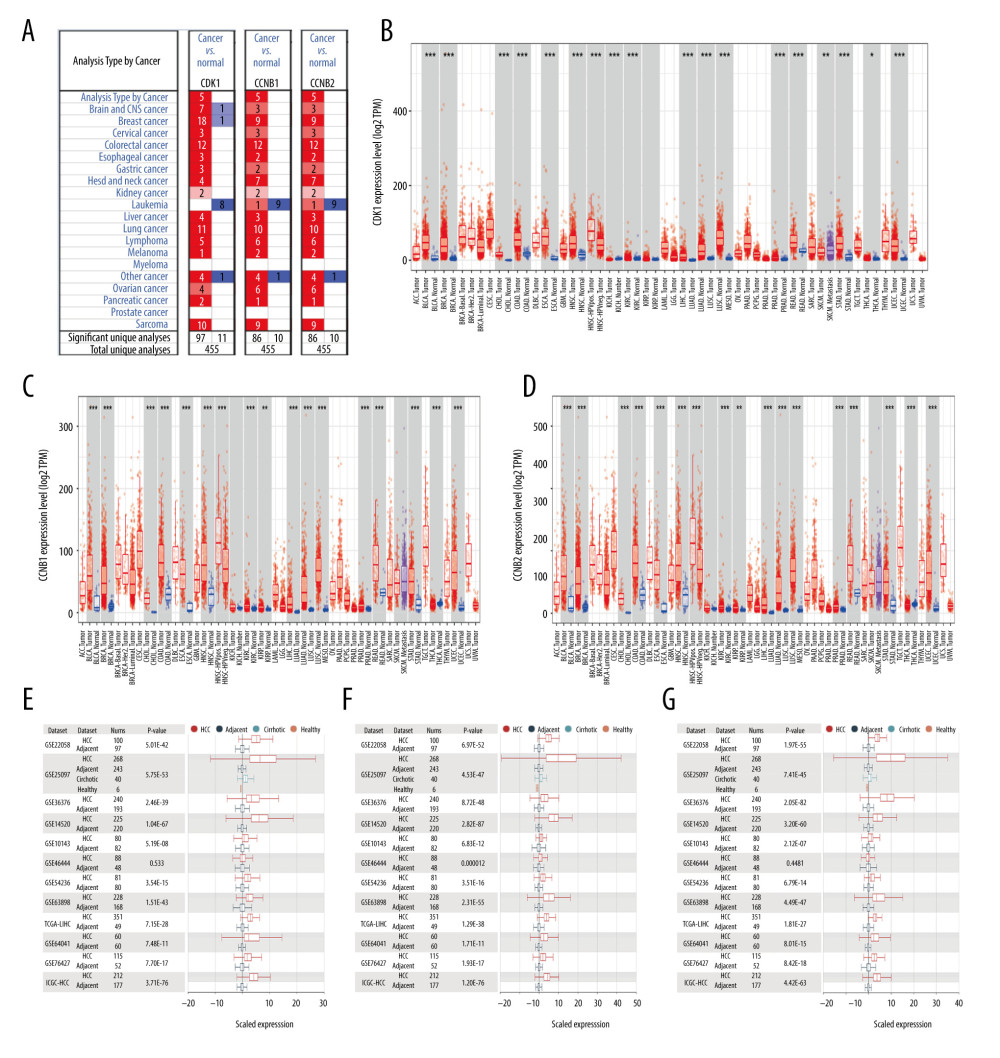 Figure 1. (A) Expression levels of CDK1, CCNB1, and CCNB2 for different types of tumors in ONCOMINE. (B–D) The levels of CDK1, CCNB1, and CCNB2 expression in different types of tumors from the TCGA database in TIMER. * P<0.05, ** P<0.01, *** P<0.001. (E–G) Charts and plots showing the expression of CDK1, CCNB1, and CCNB2 in HCC tissues and the adjacent normal tissues in HCCDB.
Figure 1. (A) Expression levels of CDK1, CCNB1, and CCNB2 for different types of tumors in ONCOMINE. (B–D) The levels of CDK1, CCNB1, and CCNB2 expression in different types of tumors from the TCGA database in TIMER. * P<0.05, ** P<0.01, *** P<0.001. (E–G) Charts and plots showing the expression of CDK1, CCNB1, and CCNB2 in HCC tissues and the adjacent normal tissues in HCCDB. 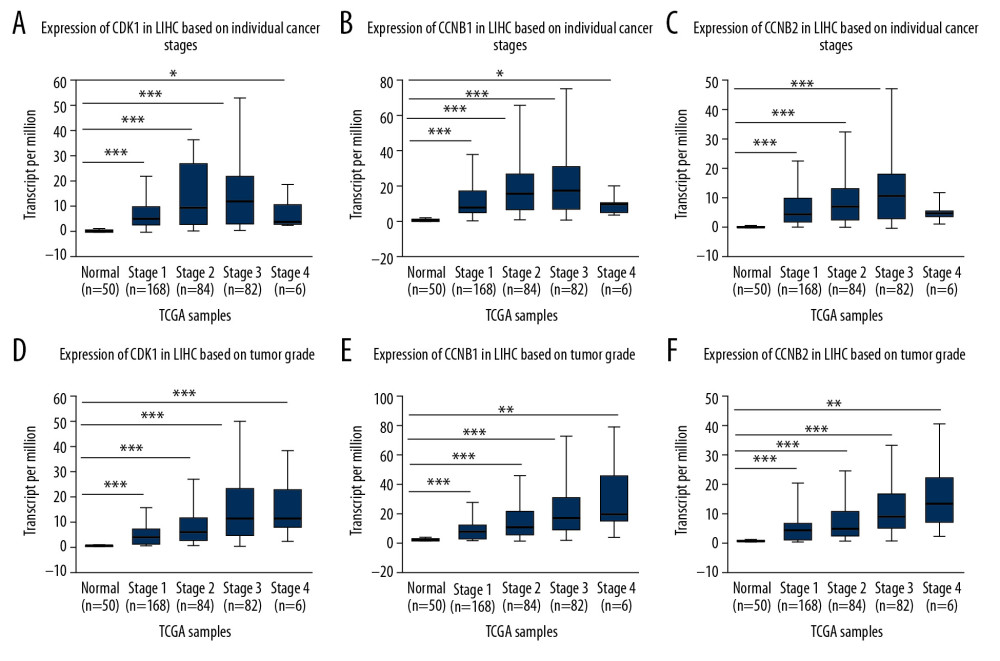 Figure 2. (A–C) Expression levels of CDK1, CCNB1, and CCNB2 in normal tissues or in HCC tissues at different stages. (D–F) Expression levels of CDK1, CCNB1, and CCNB2 in normal tissues or in HCC tissues with different grades. * P<0.05; ** P<0.01; *** P<0.001.
Figure 2. (A–C) Expression levels of CDK1, CCNB1, and CCNB2 in normal tissues or in HCC tissues at different stages. (D–F) Expression levels of CDK1, CCNB1, and CCNB2 in normal tissues or in HCC tissues with different grades. * P<0.05; ** P<0.01; *** P<0.001. 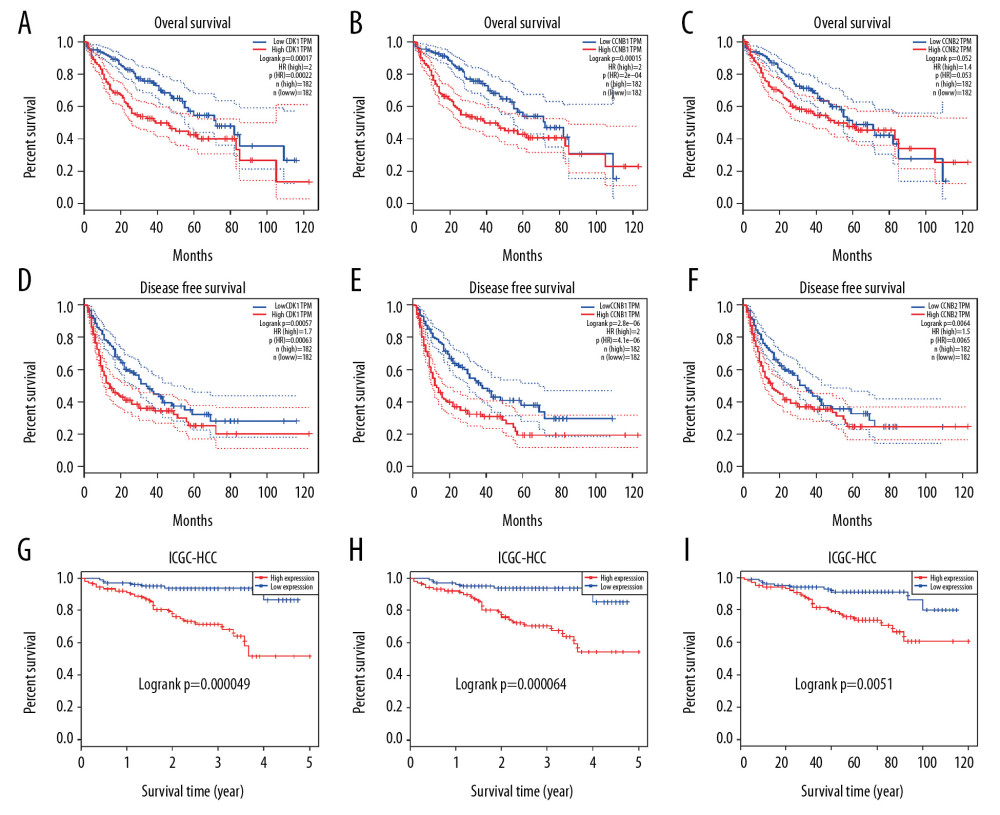 Figure 3. (A–C) Overall survival (OS) and differential CDK1, CCNB1, and CCNB2 expression in the TCGA-LIHC cohort. (D–F) Disease-free survival (DFS) and differential CDK1, CCNB1, and CCNB2 expression in the TCGA-LIHC cohort. (G–I) Overall survival (OS) and differential CDK1, CCNB1, and CCNB2 expression in the ICGC-HCC cohort.
Figure 3. (A–C) Overall survival (OS) and differential CDK1, CCNB1, and CCNB2 expression in the TCGA-LIHC cohort. (D–F) Disease-free survival (DFS) and differential CDK1, CCNB1, and CCNB2 expression in the TCGA-LIHC cohort. (G–I) Overall survival (OS) and differential CDK1, CCNB1, and CCNB2 expression in the ICGC-HCC cohort. 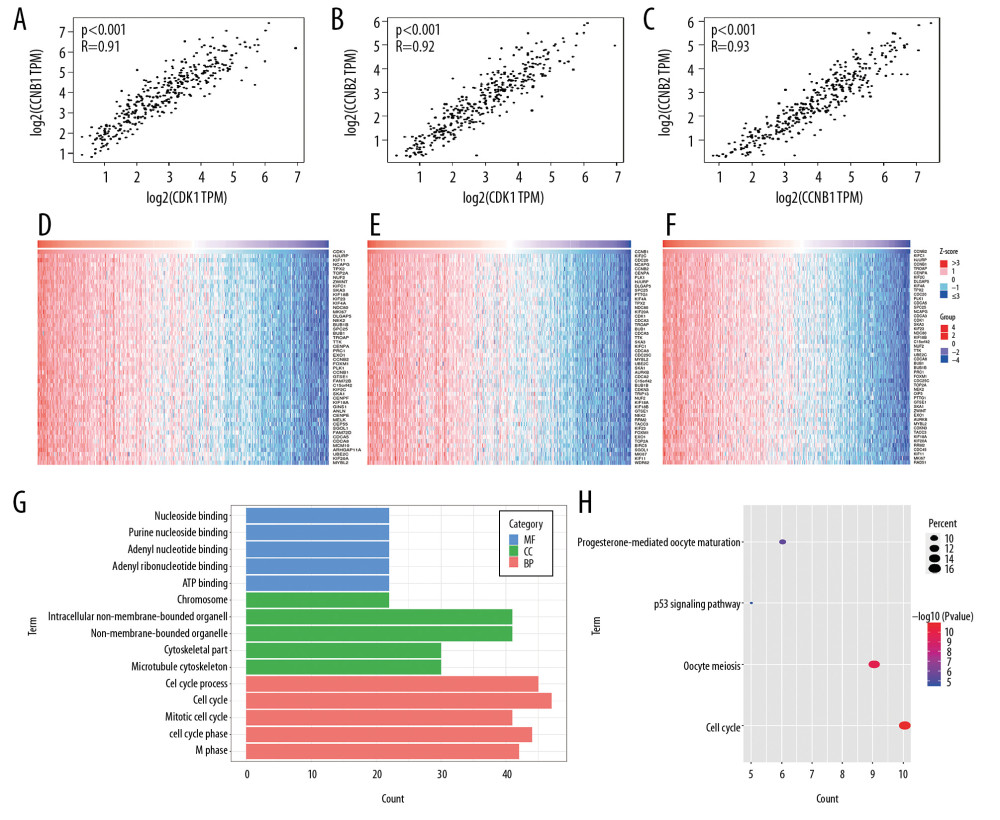 Figure 4. (A) The correlation between CDK1 and CCNB1 in HCC. (B) The correlation between CDK1 and CCNB2 in HCC. (C) The correlation between CCNB1 and CCNB2 in HCC. (D–F) Top 50 genes positively correlated with CDK1, CCNB1, and CCNB2 in LIHC from the TCGA database. (G, H) Significantly enriched GO annotations and KEGG pathways of CDK1, CCNB1, and CCNB2 in the LIHC cohort. GO – Gene Ontology; CC – cellular component; BP – biological process; MF – molecular function; KEGG – Kyoto Encyclopedia of Genes and Genomes.
Figure 4. (A) The correlation between CDK1 and CCNB1 in HCC. (B) The correlation between CDK1 and CCNB2 in HCC. (C) The correlation between CCNB1 and CCNB2 in HCC. (D–F) Top 50 genes positively correlated with CDK1, CCNB1, and CCNB2 in LIHC from the TCGA database. (G, H) Significantly enriched GO annotations and KEGG pathways of CDK1, CCNB1, and CCNB2 in the LIHC cohort. GO – Gene Ontology; CC – cellular component; BP – biological process; MF – molecular function; KEGG – Kyoto Encyclopedia of Genes and Genomes. 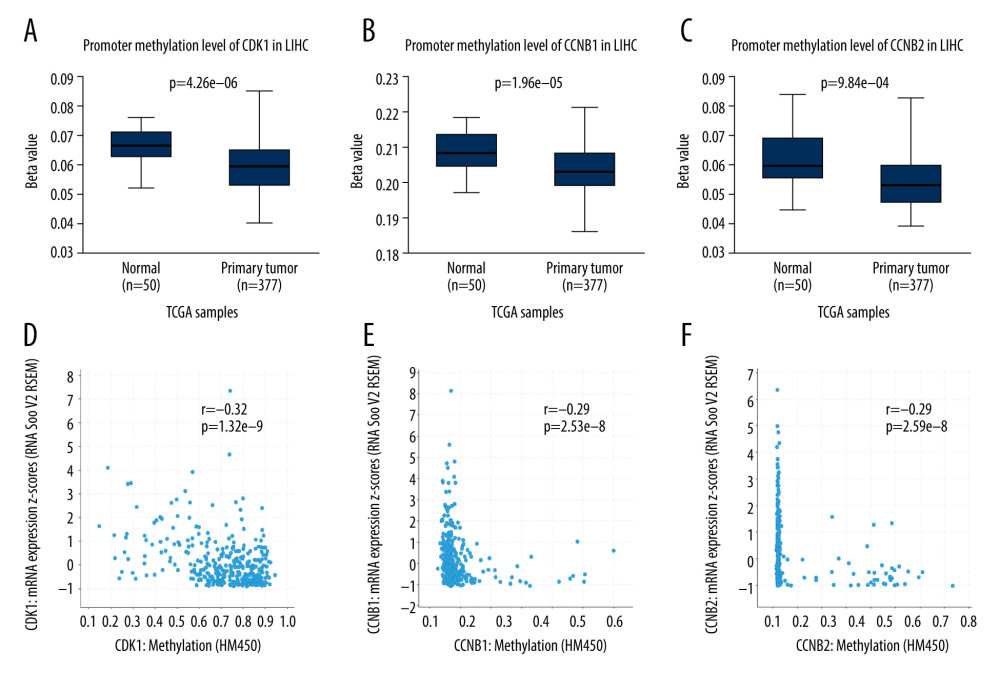 Figure 5. (A–C) Boxplots showing relative promoter methylation levels of CDK1, CCNB1, and CCNB2 in normal and LIHC samples. (D–F) The correlation between expression levels of CDK1, CCNB1, and CCNB2 and their methylation levels in LIHC samples.
Figure 5. (A–C) Boxplots showing relative promoter methylation levels of CDK1, CCNB1, and CCNB2 in normal and LIHC samples. (D–F) The correlation between expression levels of CDK1, CCNB1, and CCNB2 and their methylation levels in LIHC samples. 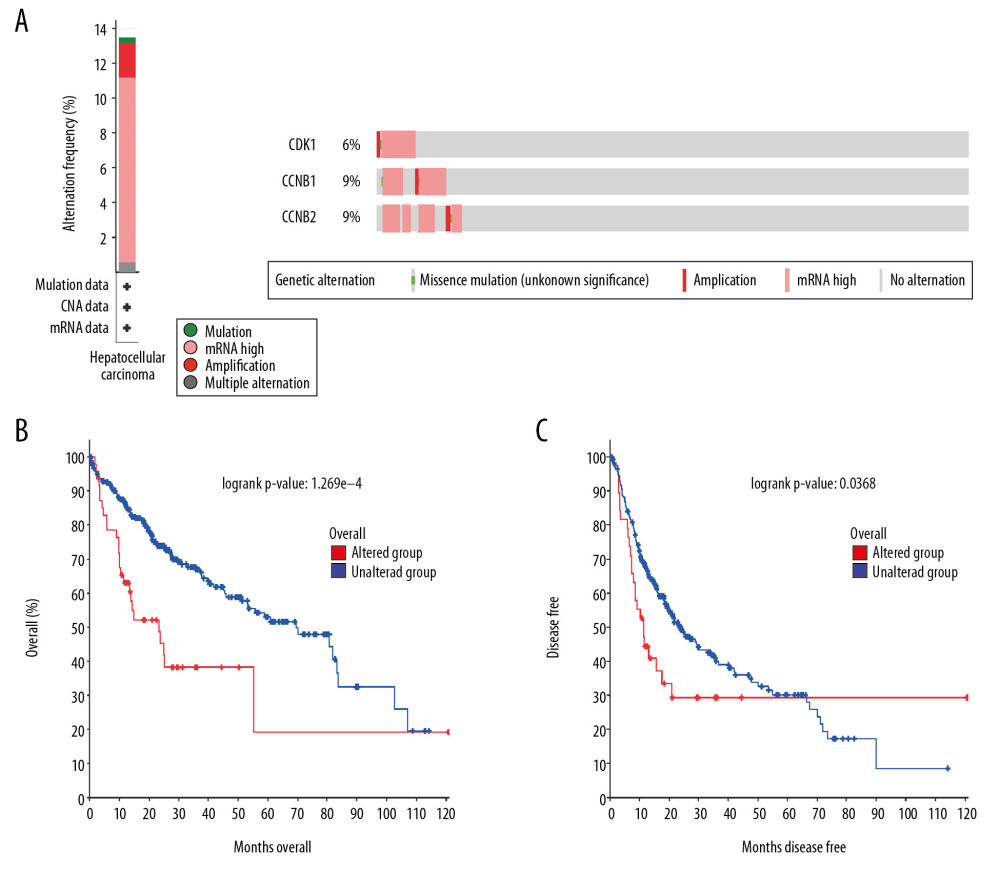 Figure 6. (A) CDK1, CCNB1, and CCNB2 mutation rates were 6%, 9%, and 9%, respectively. (B, C) Genetic alterations in CDK1, CCNB1, and CCNB2 were associated with shorter overall survival (OS) and disease-free survival (DFS) of HCC patients.
Figure 6. (A) CDK1, CCNB1, and CCNB2 mutation rates were 6%, 9%, and 9%, respectively. (B, C) Genetic alterations in CDK1, CCNB1, and CCNB2 were associated with shorter overall survival (OS) and disease-free survival (DFS) of HCC patients. 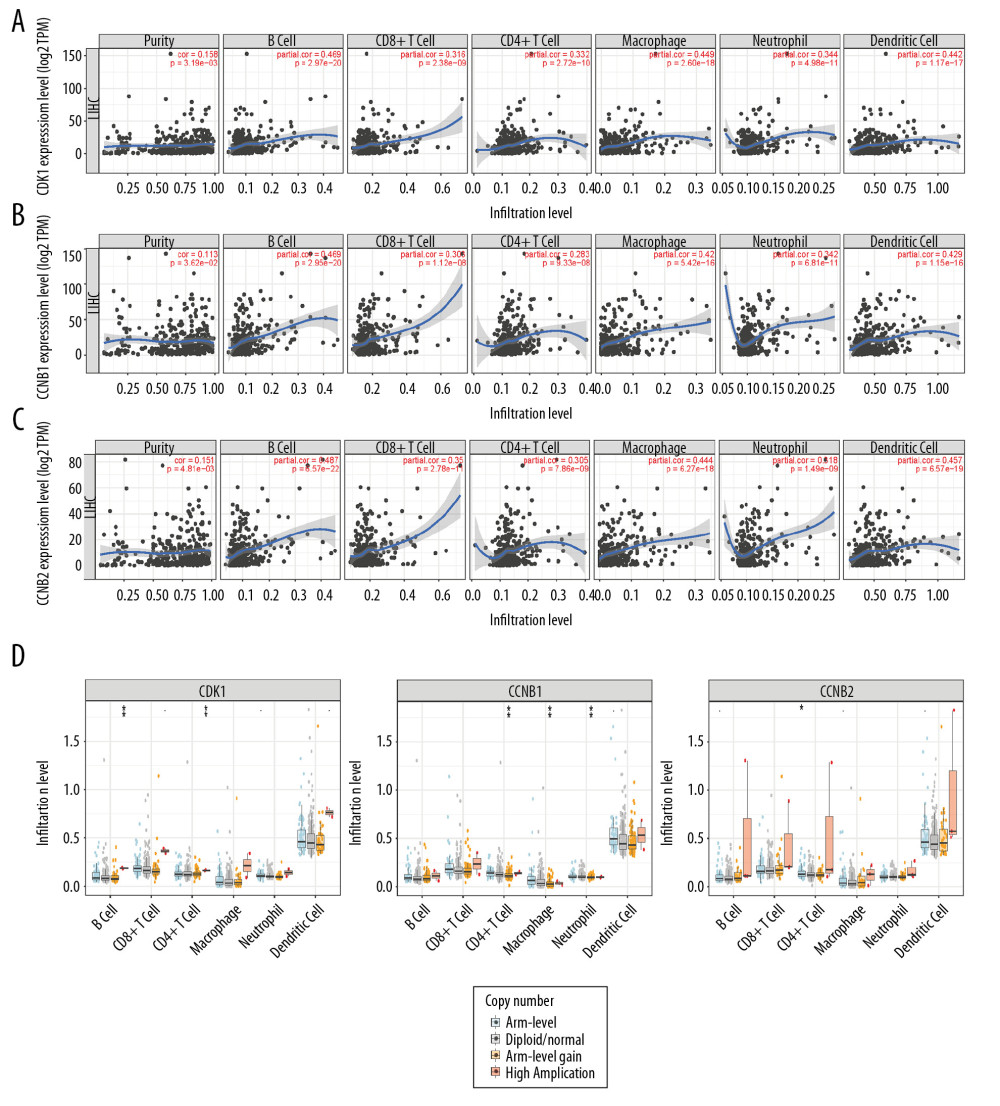 Figure 7. (A–C) CDK1, CCNB1, and CCNB2 expression levels were significantly related to tumor purity and significant positive correlations existed with immune infiltration cells including CD4+ T cells, CD8+ T cells, B cells, neutrophils, macrophages, and DCs in HCC. (D) CNA of CDK1 had significant correlations with immune infiltration cells including CD4+ T cells and B cells. CNA of CCNB1 had significant correlations with CD4+ T cells, neutrophils, and macrophages. CNA of CCNB2 had a significant correlation with CD4+ T cell. CNA, copy number alteration.
Figure 7. (A–C) CDK1, CCNB1, and CCNB2 expression levels were significantly related to tumor purity and significant positive correlations existed with immune infiltration cells including CD4+ T cells, CD8+ T cells, B cells, neutrophils, macrophages, and DCs in HCC. (D) CNA of CDK1 had significant correlations with immune infiltration cells including CD4+ T cells and B cells. CNA of CCNB1 had significant correlations with CD4+ T cells, neutrophils, and macrophages. CNA of CCNB2 had a significant correlation with CD4+ T cell. CNA, copy number alteration. References
1. Bray F, Ferlay J, Soerjomataram I, Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries: Cancer J Clin, 2018; 68(6); 394-424
2. Kew MC, Epidemiology of chronic hepatitis B virus infection, hepatocellular carcinoma, and hepatitis B virus-induced hepatocellular carcinoma: Pathol Biol (Paris), 2010; 58(4); 273-77
3. Forner A, Reig M, Bruix J, Hepatocellular carcinoma: Lancet, 2018; 391(10127); 1245-55
4. Ashhab AA, Rodin H, Powell J, Debes JD, Hepatocellular carcinoma diagnosis and surveillance: Socioeconomic factors don’t seem to matter, unless you are an immigrant: J Hepatol, 2017; 67(3); 648-49
5. Kulik L, El-Serag HB, Epidemiology and management of hepatocellular carcinoma: Gastroenterology, 2019; 156(2); 477-91
6. Cheng AL, Kang YK, Chen Z, Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: A phase III randomised, double-blind, placebo-controlled trial: Lancet Oncol, 2009; 10(1); 25-34
7. Kudo M, Finn RS, Qin S, Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: A randomised phase 3 non-inferiority trial: Lancet, 2018; 391(10126); 1163-73
8. Malumbres M, Barbacid M, Mammalian cyclin-dependent kinases: Trends Biochem Sci, 2005; 30(11); 630-41
9. Roskoski R, Cyclin-dependent protein serine/threonine kinase inhibitors as anticancer drugs: Pharmacol Res, 2019; 139; 471-88
10. Zhou J, Han S, Qian W: Onco Targets Ther, 2018; 11; 4451-59
11. Gu J, Liu X, Li J, He Y: Cancer Cell Int, 2019; 19; 15
12. Li R, Jiang X, Zhang Y, Cyclin B2 overexpression in human hepatocellular carcinoma is associated with poor prognosis: Arch Med Res, 2019; 50(1); 10-17
13. Gao X, Wang X, Zhang S, Bioinformatics identification of crucial genes and pathways associated with hepatocellular carcinoma: Biosci Rep, 2018; 38(6); BSR20181441
14. Yang WX, Pan YY, You CG: Biomed Res Int, 2019; 13; 1245072
15. Wu M, Liu Z, Li X, Analysis of potential key genes in very early hepatocellular carcinoma: World J Surg Oncol, 2019; 17(1); 77
16. Wang M, Wang L, Wu S, Identification of key genes and prognostic value analysis in hepatocellular carcinoma by integrated bioinformatics analysis: Int J Genomics, 2019; 2019 3518378
17. Rhodes DR, Yu J, Shanker K, ONCOMINE: A cancer microarray database and integrated data-mining platform: Neoplasia, 2004; 6(1); 1-6
18. Lian Q, Wang S, Zhang G, HCCDB: A database of hepatocellular carcinoma expression atlas: Genomics Proteomics Bioinformatics, 2018; 16(4); 269-75
19. Chandrashekar DS, Bashel B, Balasubramanya SA, UALCAN: A portal for facilitating tumor subgroup gene expression and survival analyses: Neoplasia, 2017; 19(8); 649-58
20. Tang Z, Li C, Kang B, GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses: Nucleic Acids Res, 2017; 45(W1); W98-102
21. Vasaikar SV, Straub P, Wang J, Zhang B, LinkedOmics: Analyzing multi-omics data within and across 32 cancer types: Nucleic Acids Res, 2018; 46(D1); D956-63
22. Modhukur V, Iljasenko T, Metsalu T, MethSurv: A web tool to perform multivariable survival analysis using DNA methylation data: Epigenomics, 2018; 10(3); 277-88
23. Gao J, Aksoy BA, Dogrusoz U, Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal: Sci Signal, 2013; 6(269); pl1
24. Li T, Fan J, Wang B, TIMER: A web server for comprehensive analysis of tumor-infiltrating immune cells: Cancer Res, 2017; 77(21); e108-10
25. Qian BZ, Pollard JW, Macrophage diversity enhances tumor progression and metastasis: Cell, 2010; 141(1); 39-51
26. Yeung OW, Lo CM, Ling CC, Alternatively activated (M2) macrophages promote tumour growth and invasiveness in hepatocellular carcinoma: J Hepatol, 2015; 62(3); 607-16
27. Najafi M, Farhood B, Mortezaee K, Contribution of regulatory T cells to cancer: A review: J Cell Physiol, 2019; 234(6); 7983-93
28. Gao Q, Qiu SJ, Fan J, Intratumoral balance of regulatory and cytotoxic T cells is associated with prognosis of hepatocellular carcinoma after resection: J Clin Oncol, 2007; 25(18); 2586-93
29. Zhang Z, Liu S, Zhang B, T cell dysfunction and exhaustion in cancer: Front Cell Dev Biol, 2020; 8; 17
30. Wirtz TH, Brandt EF, Berres ML, Liver DCs in health and disease: Int Rev Cell Mol Biol, 2019; 348; 263-99
31. Greten TF, Lai CW, Li G, Staveley-O’Carroll KF, Targeted and immune-based therapies for hepatocellular carcinoma: Gastroenterology, 2019; 156(2); 510-24
32. Stephenson JJ, Nemunaitis J, Joy AA, Randomized phase 2 study of the cyclin-dependent kinase inhibitor dinaciclib (MK-7965) versus erlotinib in patients with non-small cell lung cancer: Lung Cancer, 2014; 83(2); 219-23
33. Mitri Z, Karakas C, Wei C, A phase 1 study with dose expansion of the CDK inhibitor dinaciclib (SCH 727965) in combination with epirubicin in patients with metastatic triple negative breast cancer: Invest New Drugs, 2015; 33(4); 890-94
34. Flynn J, Jones J, Johnson AJ, Dinaciclib is a novel cyclin-dependent kinase inhibitor with significant clinical activity in relapsed and refractory chronic lymphocytic leukemia: Leukemia, 2015; 29(7); 1524-29
35. Lücking U, Jautelat R, Krüger M, The lab oddity prevails: Discovery of pan-CDK inhibitor (R)-S-cyclopropyl-S-(4-{[4-{[(1R,2R)-2-hydroxy-1-methylpropyl]oxy}-5-(trifluoromethyl)pyrimidin-2-yl]amino}phenyl)sulfoximide (BAY 1000394) for the treatment of cancer: Chem Med Chem, 2013; 8(7); 1067-85
36. Bahleda R, Grilley-Olson JE, Govindan R, Phase I dose-escalation studies of roniciclib, a pan-cyclin-dependent kinase inhibitor, in advanced malignancies: Br J Cancer, 2017; 116(12); 1505-12
37. Teo ZL, Versaci S, Dushyanthen S, Combined CDK4/6 and PI3Kα inhibition is synergistic and immunogenic in triple-negative breast cancer: Cancer Res, 2017; 77(22); 6340-52
38. De Luca A, Maiello MR, D’Alessio A, Pharmacokinetic drug evaluation of palbociclib for the treatment of breast cancer: Expert Opin Drug Metab Toxicol, 2018; 14(9); 891-900
Figures
Figure 1. (A) Expression levels of CDK1, CCNB1, and CCNB2 for different types of tumors in ONCOMINE. (B–D) The levels of CDK1, CCNB1, and CCNB2 expression in different types of tumors from the TCGA database in TIMER. * P<0.05, ** P<0.01, *** P<0.001. (E–G) Charts and plots showing the expression of CDK1, CCNB1, and CCNB2 in HCC tissues and the adjacent normal tissues in HCCDB.Figure 2. (A–C) Expression levels of CDK1, CCNB1, and CCNB2 in normal tissues or in HCC tissues at different stages. (D–F) Expression levels of CDK1, CCNB1, and CCNB2 in normal tissues or in HCC tissues with different grades. * P<0.05; ** P<0.01; *** P<0.001.Figure 3. (A–C) Overall survival (OS) and differential CDK1, CCNB1, and CCNB2 expression in the TCGA-LIHC cohort. (D–F) Disease-free survival (DFS) and differential CDK1, CCNB1, and CCNB2 expression in the TCGA-LIHC cohort. (G–I) Overall survival (OS) and differential CDK1, CCNB1, and CCNB2 expression in the ICGC-HCC cohort.Figure 4. (A) The correlation between CDK1 and CCNB1 in HCC. (B) The correlation between CDK1 and CCNB2 in HCC. (C) The correlation between CCNB1 and CCNB2 in HCC. (D–F) Top 50 genes positively correlated with CDK1, CCNB1, and CCNB2 in LIHC from the TCGA database. (G, H) Significantly enriched GO annotations and KEGG pathways of CDK1, CCNB1, and CCNB2 in the LIHC cohort. GO – Gene Ontology; CC – cellular component; BP – biological process; MF – molecular function; KEGG – Kyoto Encyclopedia of Genes and Genomes.Figure 5. (A–C) Boxplots showing relative promoter methylation levels of CDK1, CCNB1, and CCNB2 in normal and LIHC samples. (D–F) The correlation between expression levels of CDK1, CCNB1, and CCNB2 and their methylation levels in LIHC samples.Figure 6. (A) CDK1, CCNB1, and CCNB2 mutation rates were 6%, 9%, and 9%, respectively. (B, C) Genetic alterations in CDK1, CCNB1, and CCNB2 were associated with shorter overall survival (OS) and disease-free survival (DFS) of HCC patients.Figure 7. (A–C) CDK1, CCNB1, and CCNB2 expression levels were significantly related to tumor purity and significant positive correlations existed with immune infiltration cells including CD4+ T cells, CD8+ T cells, B cells, neutrophils, macrophages, and DCs in HCC. (D) CNA of CDK1 had significant correlations with immune infiltration cells including CD4+ T cells and B cells. CNA of CCNB1 had significant correlations with CD4+ T cells, neutrophils, and macrophages. CNA of CCNB2 had a significant correlation with CD4+ T cell. CNA, copy number alteration. Tables
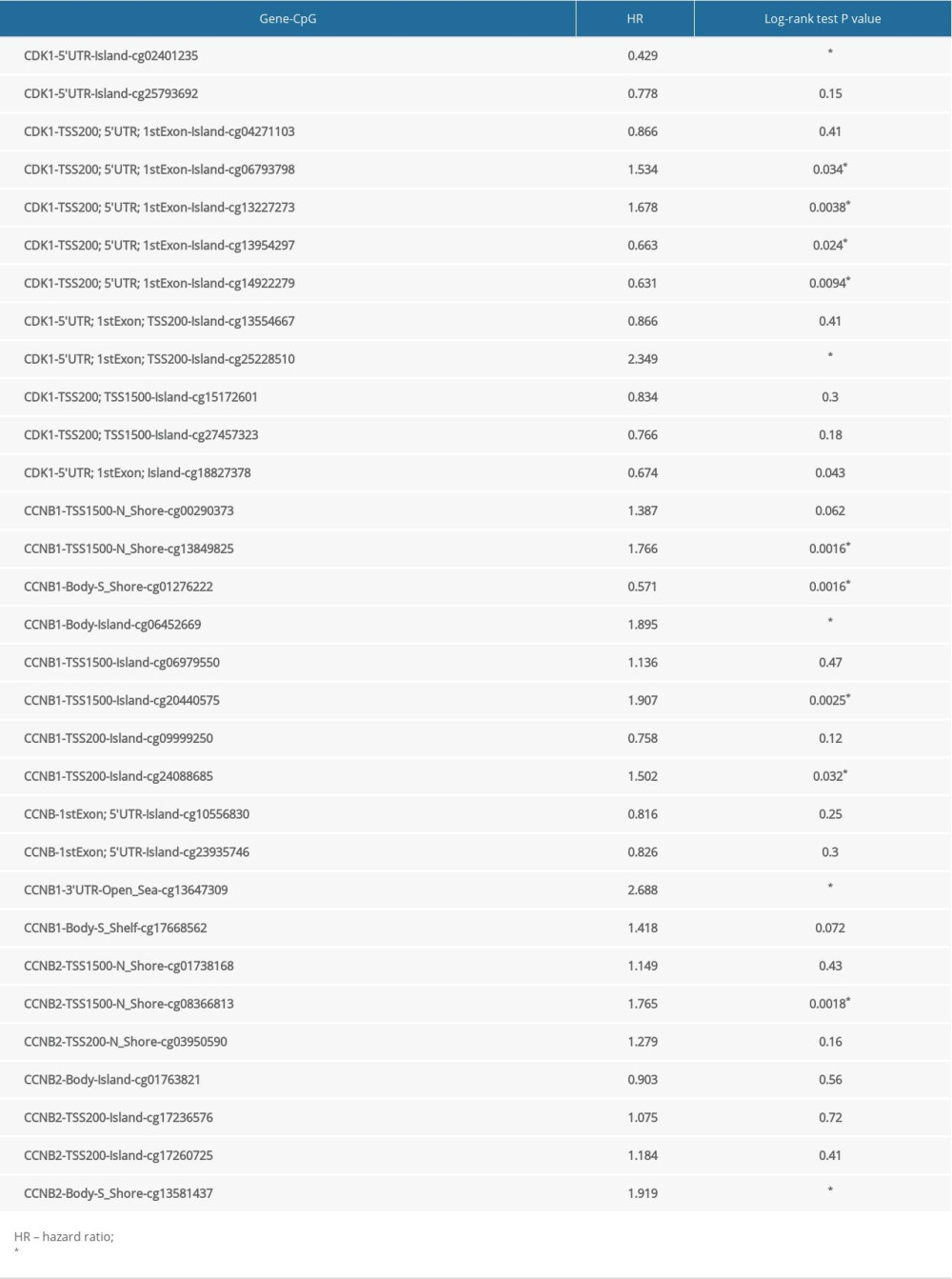 Table 1. The prognostic values of CpG in CDK1, CCNB1, and CCNB2 genes by MethSurv.
Table 1. The prognostic values of CpG in CDK1, CCNB1, and CCNB2 genes by MethSurv.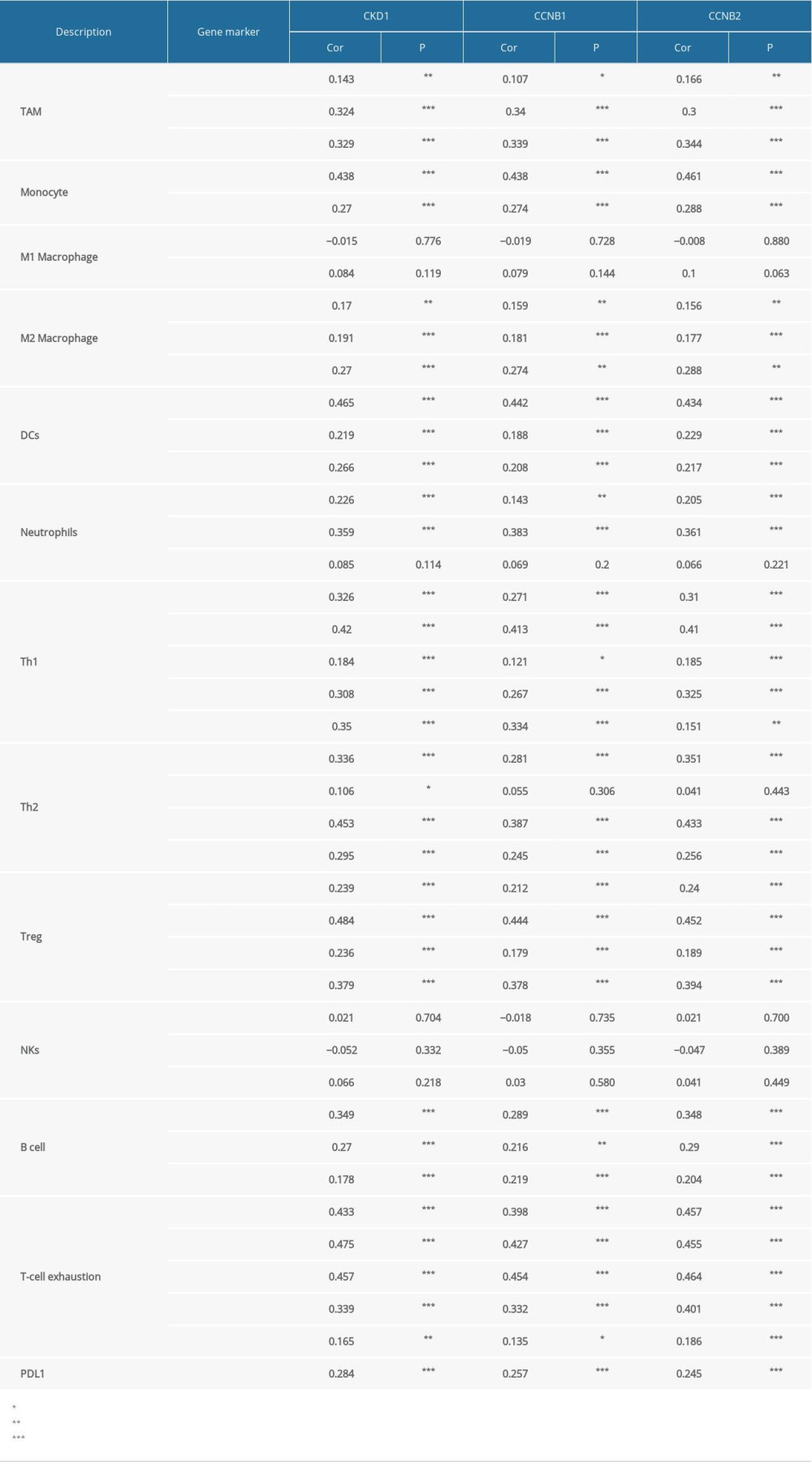 Table 2. Correlation analysis between CDK1, CCNB1, CCNB2, and related immune markers.Table 1. The prognostic values of CpG in CDK1, CCNB1, and CCNB2 genes by MethSurv.Table 2. Correlation analysis between CDK1, CCNB1, CCNB2, and related immune markers.
Table 2. Correlation analysis between CDK1, CCNB1, CCNB2, and related immune markers.Table 1. The prognostic values of CpG in CDK1, CCNB1, and CCNB2 genes by MethSurv.Table 2. Correlation analysis between CDK1, CCNB1, CCNB2, and related immune markers. In Press
Clinical Research
Effects of Single-Bout Endurance Exercise Intensity on Peripheral Neurotrophic Factors in Patients With Isc...Med Sci Monit In Press; DOI: 10.12659/MSM.952089
Review article
Anisodus tanguticus in Cancer Research: A Review of Traditional Use, Phytochemistry, Extraction Methods, an...Med Sci Monit In Press; DOI: 10.12659/MSM.952999
Clinical Research
Nasal Mucociliary Clearance and Its Relationship With Disease Severity in Patients With Multiple SclerosisMed Sci Monit In Press; DOI: 10.12659/MSM.952850
Clinical Research
Modified Thoracoabdominal Nerves Block Through the Perichondrial Approach vs Subcostal Transversus Abdomini...Med Sci Monit In Press; DOI: 10.12659/MSM.953976
Most Viewed Current Articles
17 Jan 2024 : Review article 14,176,570
Vaccination Guidelines for Pregnant Women: Addressing COVID-19 and the Omicron VariantDOI :10.12659/MSM.942799
Med Sci Monit 2024; 30:e942799
13 Nov 2021 : Clinical Research 3,762,188
Acceptance of COVID-19 Vaccination and Its Associated Factors Among Cancer Patients Attending the Oncology ...DOI :10.12659/MSM.932788
Med Sci Monit 2021; 27:e932788
14 Dec 2022 : Clinical Research 2,466,310
Prevalence and Variability of Allergen-Specific Immunoglobulin E in Patients with Elevated Tryptase LevelsDOI :10.12659/MSM.937990
Med Sci Monit 2022; 28:e937990
16 May 2023 : Clinical Research 708,927
Electrophysiological Testing for an Auditory Processing Disorder and Reading Performance in 54 School Stude...DOI :10.12659/MSM.940387
Med Sci Monit 2023; 29:e940387