06 April 2021: Review Articles
Impact of Cardiovascular Diseases on COVID-19: A Systematic Review
Qingtai Cao1B*, HanYu Lei23E, MengLing Yang23B, Le Wei23A, YinMiao Dong2E, JiaHao Xu23F, Mi Nasser4C, MengQi Liu23F, Ping Zhu4G, LinYong Xu5G, MingYi Zhao2AGDOI: 10.12659/MSM.930032
Med Sci Monit 2021; 27:e930032






Abstract
ABSTRACT: In December 2019, pneumonia of unknown cause broke out, and currently more than 150 countries around the world have been affected. Globally, as of 5: 46 pm CET, 6 November 2020, the World Health Organization (WHO) had reported 48 534 508 confirmed cases of COVID-19, including 1 231 017 deaths. The novel coronavirus disease (COVID-19) outbreak, caused by the SARS-CoV-2 virus, is the most important medical challenge in decades. Previous research mainly focused on the exploration of lung changes. However, with development of the disease and deepening research, more and more patients showed cardiovascular diseases, even in those without respiratory symptoms, and some researchers have found that underlying cardiovascular diseases increase the risk of infection. Although the related mechanism is not thoroughly studied, based on existing research, we speculate that the interaction between the virus and its receptor, inflammatory factors, various forms of the stress response, hypoxic environment, and drug administration could all induce the development of cardiac adverse events. Interventions to control these pathogenic factors may effectively reduce the occurrence of cardiovascular complications. This review summarizes the latest research on the relationship between COVID-19 and its associated cardiovascular complications, and we also explore possible mechanisms and treatments.
Keywords: Cardiovascular Diseases, Cell Hypoxia, COVID-19, Stress, Physiological, Systemic Inflammatory Response Syndrome, COVID-19, Pandemics, SARS-CoV-2, World Health Organization
Summary
Globally, as of 5: 46 pm CET, 6 November 2020, there have been 48 534 508 confirmed cases of COVID-19, including 1 231 017 deaths, as reported by to the WHO [1]. Infected patients have similar clinical symptoms as those who have viral pneumonia [2]. After the isolation and identification of the virus, the pneumonia pathogen was initially called the 2019 new coronavirus (2019-nCoV), and then the WHO officially named it severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2).
In the initial report, Huang et al found that about 12% of patients with COVID-19 were diagnosed with acute myocardial injury [2]. In another cohort study of patients with confirmed COVID-19, cardiac injury occurred in 19.7%, and it has become an independent risk factor for in-hospital mortality [3]. A study of patients with severe symptoms of COVID-19 found that 58% had hypertension, 25% had heart disease, and 44% had arrhythmia [4]. Ruan et al identified clinical predictors of mild and severe patient outcomes based on an analysis of 150 patients from Wuhan, China. They noted that patients with cardiovascular diseases have a significantly increased risk of death when they are infected with SARS-CoV-2 (
Related Mechanisms
CARDIOVASCULAR SYSTEM DAMAGE CAUSED BY BINDING TO ACE2:
It has been suggested that SARS-CoV-2 infection is associated with angiotensin-converting enzyme 2 (ACE2) [7,8]. ACE2 is an important peptidase in the renin-angiotensin system (RAS), degrading Ang I into its inactive form, Ang 1–9. It can also hydrolyze Ang II, which promotes cardiovascular diseases, into a potent vasodilator, Ang (1–7). Recently, ACE2 was verified as the functional receptor for coronaviruses [9]. The virus can directly damage cells in the cardiovascular system by entry into host cells expressing ACE2. Given the essential nature of ACE2 for viral infection, the distribution of ACE2 expression is informative to elucidate the likely infected tissues and hypothetical mechanisms of injury. ACE2 has been demonstrated to be widely expressed in multiple tissues and organs such as the lungs, kidneys, small intestine, brain, and cardiovascular system. As the receptor of SARS-CoV-2, the data on ACE2 expression indicate that the existence of ACE2 might facilitate SARS-CoV-2 invasions outside the lungs. Recently, Sharma et al [10] revealed that SARS-CoV-2 could invade cardiomyocytes derived from a pluripotent stem cell in vitro via ACE2, leading to increased cell apoptosis and decreased ACE2 expression. This finding suggests that cardiovascular complications in COVID-19 patients might be a direct or indirect consequence of the interactions of virus binding and ACE2 expressed within the cardiovascular system. A single-cell atlas of the human heart revealed high and specific expression on pericytes that have angiopoietin ligands (ANGPT1/2)–Tie receptor (TIE2) crosstalk with capillary endothelial cells, thus affecting the function of capillary vessels [11], indicating SARS-CoV-2 might cause micro-circulation disorder via ACE2 on pericytes. Also, SARS-CoV-2 can lead to downregulation of ACE2 through several processes. During the viral entry process, when the N-terminal portion of the viral protein unit S1 binds to a pocket of the ACE2 receptor, the virus can activate tumor necrosis factor α-converting enzyme (TACE/ADAM17) and cause cleavage of ACE2 protein and shedding of its extracellular domain [12,13]. Subsequently, the virions appear to cause a subsequent membrane fusion, resulting in entry into host cells, along with the membrane receptor [14]. As a result, ACE2 is functionally removed from the external site of the membrane. As with transcriptional downregulation of receptor mRNA induced by cytokines in multiple viral infections including HIV, measles, and SARS-CoV [15], SARS-CoV-2 might also lead to a downregulated level of ACE2 mRNA through a similar mechanism. The rapid downregulation of ACE2 resulted in the enhanced effect of Ang II, the absence of cardioprotective effect of Ang 1–7, and the sustained activation of ADAM-17/TACE. The overexpression of angiotensin II can lead to multiple adverse outcomes such as cardiac hypertrophy [16], disorders in blood pressure regulation, and worsening problems with oxidative stress, inflammation, and coagulation [17]. Excessive expression of angiotensin II can stimulate the production of proinflammatory cytokines such as IL-6 [18] and TNF-α [19] by accelerating macrophages polarizing into M1 phenotypes or activating other immunocytes. In addition to RAS disorders, the downregulation of ACE2 is also responsible for continuous activation of ADAM-17/TACE, a high level of which can stimulate TNA-α expression, thus aggravating the inflammatory injury in cardiovascular tissues. This can partly explain the increased risk of cardiovascular complications, especially heart failure and venous thromboembolic events [20], and the cardiovascular system findings from autopsy of patients who died due to SARS-CoV-2 [21].
The downregulation of ACE2 in other organs or tissues outside the cardiovascular system can also disturb cardiovascular function. According to the relatively high incidence of neurological symptoms in COVID-19 patients and a recent finding in brain biopsies that SARS-CoV-2 RNA was detected in 36.4% (8/22) of COVID-19 cases [22], the virions are likely to infect the central nervous system (CNS) directly. Successful infection and replication of SARS-CoV-2 in human neuronal origin cells further revealed this possibility [23]. Since ACE2 expressed in the hypothalamus has been revealed to have antihypertensive and sympatholytic abilities [24], virus-induced receptor decrease might facilitate an enhanced sympathetic drive and altered baroreflex, thus causing cardiovascular system damage. A similar alteration of ACE2 expression might occur in the kidney as well, where the loss of ACE2 on tubular epithelium contributes to an alteration of sodium transport, inducing a relatively high blood volume and blood pressure to increase the cardiovascular load. In addition, the low level of ACE2 in lung tissues might aggravate hypertension secondary to viral infection [25]. The possible mechanisms of SARS-CoV-2-related cardiovascular complications induced by the downregulation of ACE2 are shown in Figure 1.
STRESS:
The manifestation of acute cardiac injuries such as enhanced cardiac troponin and abnormal electrocardiographic appear to be prevalent in hospitalized COVID-19 patients, among which the level of cardiac troponin is reported to be correlated with a higher risk of death [3,26]. A high level of cardiac troponin could be a result of oxidative stress caused by hypoxia, accumulated proinflammatory cytokines, or alteration of ACE2 expression. The downregulation of ACE2 by SARS-CoV-2 could be a potential activator of oxidative stress in the cardiovascular system, as the accumulation of angiotensin II can activate Nox2, which is a key enzyme in reactive oxidant species (ROS) generation [27] and has been revealed to be upregulated in pneumonia [28]. In addition, the level of NoX2 is independently correlated with heart failure [29] and troponin elevation [27], suggesting that activation of Nox2 is a possible cause of cardiovascular complications in COVID-19.
Several species of respiratory viruses, such as influenza and respiratory syncytial virus, have been reported to induce the formation of ROS within cells or at sites with excessive inflammatory cell recruitment via inhibiting Nrf2 pathways or activating NF-κB signaling [30]. In human CoV (HCoV) infection, Shao et al reported an increase in the expression of the genes related to oxidative stress in peripheral blood mononuclear cells, as well as those with high sensitivity to oxidative stress [31]. Since the cardiovascular cells might be target cells of SARS-CoV-2, the virus-related upregulation of oxidative stress could be one of the causes of damage to heart tissues. As a main source of ROS, mitochondria are also closely related to the level of oxidative stress. The abundance of TNF-α and IL-6 existing in the serum of COVID-19 patients can interfere with oxidative phosphorylation in mitochondria, along with the production of associated ATP and ROS [32]. Mitochondrial oxidative stress has recently been demonstrated to play an important role in the development of COVID-19 [33]. The abundance of mitochondria in cardiomyocytes makes them vulnerable to pathogenic cytokines that can disturb normal physiological functions of mitochondria. Similar conjectures can even be extended to another organelle, the endoplasmic reticulum.
Endoplasmic reticulum stress is involved in the physiological process in the respiratory system, including antioxidant effects, inflammatory responses [34], and apoptosis of type II alveolar epithelial cells [35]. In light of the level of systemic oxidative stress and proinflammatory cytokines and the damage in lung cells during SARS-CoV-2 infection, there must be a high level of ER stress. The existence of ER stress was also revealed to accelerate multiple cardiac adverse events such as cardiomyocyte apoptosis, heart failure, and atherosclerosis [36]. Ying et al [37] reported that ER stress could elevate the expression levels of GM-CSF and the binding of NF-κB and the GM-CSF gene in mice. Smooth muscle cells along with the collagen in atherosclerotic plaque were also suppressed by ER stress and were restored after treatment of ER stress blockage in this research, indicating ER stress can destabilize atherosclerotic plaque partly through increasing NF-κB-induced GM-CSF, which means, with a relatively high level of GM-CSF expression, COVID-19 patients with atherosclerosis are more likely to develop atherosclerotic plaque rupture through ER stress.
HYPOXIA:
As reported before, alveolar epithelial cells that express ACE2 are the main targets in SAR-CoV-2 infection, and the main manifestations are respiratory dysfunctions such as dyspnea and dry cough. Respiratory system injuries, cytokine storm, and pulmonary shunt caused by SARS-CoV-2-induced vasodilatation [38] all contribute to a high risk of hypoxemia in COVID-19 patients, thus placing the cardiovascular tissues in a relatively hypoxic condition. Myocardial hypoxia can cause cardiovascular damage through a variety of mechanisms, one of which is enhancing the production of IL-6. Chouvarine et al [39] performed a differential qPCR analysis and RNA expression screen on cardiomyocytes from mice, which were treated with normoxia and hypoxia, and observed a lower level of miR-146b in the hypoxia group, and incubation of cardiomyocytes led to upregulated expression of TRAF6, IL-6, and CCL2 (MCP-1). These findings suggest that SARS-CoV-2-related hypoxemia can trigger cardiac dysfunction and even heart failure by affecting the miR-146b-TRAF6-IL-6/CCL2(MCP-1) axis. Hypoxia-induced mitogenic factor (HIMF), which is a cytokine-like protein initially found in hypoxia-induced pathological conditions in lung tissues [40], could be another important proinflammatory mediator in cardiovascular injuries in COVID-19. Overexpressed HIMF was observed to activate MAPK (mitogen-activated protein kinase) and CaMKII (Ca2+/calmodulin-dependent protein kinase II)-STAT3 (signal transducers and activators of transcription 3) pathways, which were related with cardiomyocyte-to-fibroblast ratio in cardiac tissues in mouse models; these pathways can be inhibited under the condition of IL-6 depletion [41]. These findings indicate that hypoxia can induce HIMF and subsequently upregulate IL-6, a downstream signal, to induce cardiomyocyte hypertrophy or fibrosis via MAPK or CaMKII-STAT3 pathways. Although there is a lack of data on the expression level of those factors in patients infected with SARS-CoV-2, hypoxia caused by pathological changes of the lung, increased IL-6, and short-term or long-term cardiovascular complications in those patients indicate the axis of HIMF could be a potential mechanism in inducing cardiovascular adverse events in COVID-19 patients.
INFLAMMATORY FACTORS:
Accumulation of cytokines and cascade amplification of inflammatory mediators are the main factors that aggravate fatal SARS-CoV-2 infections. An enhanced level of IL-6 was demonstrated to be directly correlated with the prognosis of patients with COVID-19. As an upstream inflammatory marker, IL-6 was previously evidenced to participate in the pathogenesis of cardiac dysfunctions and to be correlated with the risk of adverse cardiovascular events such as myocardial infarction and heart failure [42], which also appear to be the main cardiovascular complications in SARS-CoV-2 infection. It was documented that, compared to wild-type mice, IL-6-deficient mice had reduced incidence of infarction after ischemia-reperfusion treatment [43], indicating IL-6 participates in the formation of infarction when the tissue experiences ischemia. IL-6 also plays a vital role in myocardial remodeling. Previous findings of in vitro experiments on mouse cardiac tissues demonstrated that overexpression of IL-6 could activate TGF-β1 signaling to reprogram the processes of proliferation and/or fibrosis in fibroblasts in hypoxia conditions, thus remodeling the myocardial construction. Similar findings were observed in vivo as well. In an experiment by Jing et al in mice, model mice suffered from myocardial infarction (MI), and knockout of IL-6 alleviated TNF-α level as well as the degree of cardiac fibrosis, but increased the number of M2 macrophages after MI [44]. Those findings indicate that the acute IL-6 enhancement in COVID-19 patients might induce long-term adverse events in the cardiovascular system, which deserves more attention.
However, contrary to the proinflammatory effects, IL-6 also has a protective function in some cases, which could partly explain why the blockage of IL-6 expression did not always produce a curative effect but instead triggered proinflammatory adverse outcomes. Sanmarco and colleagues [45] surprisingly found that IL-6 affected the response of innate immunity by regulating inflammasome activation and consequently suppressed the over-production of nitric oxide, which contributed to lethal oxidative stress in myocardial tissue by inhibiting IL-1β. The promotive effects of IL-6 on lipolysis and fatty acid oxidation also make it an important mediator to protect cardiac tissues from lipotoxicity [46], which was shown to elicit endoplasmic reticulum stress or mitochondria dysfunction, thus impairing cardiac tissues [47].
In addition to the high expression of IL-6, other proinflammatory factors such as IL-17, MCP-3, TNF, and GM-CSF appear to be enhanced in COVID-19 patients [48]. As the most efficient support for COVID-19 patients with severe and automatic circulatory and respiratory dysfunction, ECMO (extracorporeal membrane oxygenation) has been used for the most serious cases [49,50] and further enlarged the systemic inflammatory response [51]. On the basis of the studies on SARS-CoV, the homologous virus of SARS-CoV-2, neutrophilia was reduced in complement-deficient mice along with systemic inflammation [52], indicating complement activation might be another mechanism for excessive cytokine production in coronavirus infections. All of those cytokines contribute to a proinflammatory environment, which is related to fatal outcomes in COVID-19 patients and induce cardiac dysfunction [53].
DRUG-RELATED INJURIES:
There is currently no specific medication for use in COVID-19 patients. Treatment generally uses antimalarial drugs, antiviral drugs, and antibacterial drugs. Chloroquine is an antimalarial drug. In vitro studies have shown that chloroquine is effective against several viruses, including coronaviruses, by impairing the replication of viruses and reducing the release of proinflammatory factors [54]. However, there has been no analysis of the clinical effect of chloroquine treatment. It was reported that chloroquine and its related formulations might prevent infection or the development of severe symptomatic disease, substantially reducing morbidity and mortality due to COVID-19 [55]. Chloroquine is ion-active, blocking IKr potassium (and other) channels [56]. This pharmacologic action may cause life-threatening arrhythmias, notably torsade de pointes (TdP) [56].
Wang et al found that most patients (89.9%) with COVID-19 have received antiviral drugs, such as oseltamivir [4], but many antiviral drugs can cause cardiac insufficiency, arrhythmia, or other cardiovascular disorders [7]. The most frequently used antiviral drugs are remdesivir, lopinavir/ritonavir, and favipiravir. The main documented adverse effect of lopinavir/ritonavir therapy in the cardiovascular system is a higher risk of atherosclerosis progression, manifested as an increased level of total cholesterol and low-density lipoprotein (LDL) and a decreased level of high-density lipoprotein (HDL) [57]. Atrioventricular blocks, sinus arrest [58], and QT and PR prolongation in electrocardiogram [20] also can occur after lopinavir/ritonavir treatment. According to the published literature on remdesivir administration, there was 1 death related to cardiac arrest or possible ebolavirus infection [59].
Azithromycin is an antibacterial medication verified by clinical trials to effectively reinforce the antiviral effects of hydroxychloroquine in COVID-19 [60] and showed certain cardiotoxic effects in previous research. Long-term administration of azithromycin is believed to increase the risk of ventricular arrhythmias via increasing Na+ current and blocking the outward current of K+ from ventricular myocytes [61]. The FDA issued a statement that azithromycin can lead to lethal arrhythmia in patients with cardiovascular disease [62], so the use of azithromycin might endanger COVID-19 patients with hypokalemia.
Quite a few newly studied medications can also generate adverse events in the cardiovascular system. Pegylated interferon-α was reported to be associated with potential cardiotoxicity, which was likely mediated by TNF-α [63] to induce cardiac complications such as pericardial effusion, cardiomyopathy, and arrhythmias [64]. High-dose vitamin C, favipiravir, ARBs, and monoclonal antibodies are undergoing clinical trials [63].
The possible mechanisms for cardiovascular complications associated with multiple types of stress, hypoxia, accumulation of inflammatory cytokines, and drug administration in COVID-19 patients are shown in Figure 2.
Related Cardiovascular Diseases
ACUTE CARDIAC INJURY:
This new virus belongs to the same family as severe acute respiratory syndrome-coronavirus (SARS-CoV) and the Middle East respiratory syndrome-coronavirus (MERS-CoV). Previous studies have found that SARS-CoV and MERS-CoV both can cause cardiac injury [65–68]. The similarity among these 3 viruses suggests that SARS-CoV-2 can cause acute heart damage, including myocardial injury and heart failure.
A study by Shi et al suggested that the cardiac injury was a predictor of death, and they reported a 19.7% incidence rate of cardiac injury [3]. In the initial report in Wuhan, China, 5 of the first 41 patients diagnosed with COVID-19 had SARS-CoV-2-related myocardial injury, and 4 of these 5 patients were admitted to the intensive-care unit (ICU) [2]. In a later published clinical cohort study of patients with COVID-19, Wang et al observed that acute cardiac injury was present in 7.2% of patients [4]. Myocardial injury biomarker levels in patients requiring ICU admission were significantly higher, which indicates that patients with severe symptoms usually have complications of acute myocardial injury [4]. Ruan et al analyzed 150 patients from Wuhan, China, and found among the 68 fatal cases, 5 patients (7%) with myocardial damage died of circulatory failure [5].
A case report published in March 2020 showed that blood tests revealed elevated levels of markers of myocyte necrosis, such as high-sensitivity troponin T (TnT), creatine kinase–MB (CK-MB), N-terminal pro-brain natriuretic peptide (NT-proBNP), and C-reactive protein [68] (Figure 3). Laboratory results also showed that there were significant differences between patients who died and those who were discharged; the levels of myocardial injury markers in the group of patients who died were significantly higher [5]. Guzik et al summarized existing research and drew some conclusions regarding diagnostic tests. For example, high concentrations of IL-6 are associated with adverse outcomes. Ferritin is a marker of poor outcome, and it has very significant changes reported in COVID-19 patients (Figure 1). Higher NT-proBNP levels in the Chinese cohort are associated with a greater need for ICU care. The high-sensitivity troponin assay may be helpful for risk assessment in patients requiring ICU care and to identify individuals with silent myocardial injury [69].
Andrea et al discovered that the frequencies of myocardial injury ranged from 3.3% to 44.4% [70]; 8 studies, with a total of 1229 patients, were included. Interestingly, there was high heterogeneity among the studies (92%). Different studies have different definitions of acute myocardial injury. Of note, the timing of the troponin measurements was different among the studies, and the patient severity was also variable. These factors may explain the different reported frequencies of myocardial injury among studies.
The mechanisms underlying acute cardiac injury remain unknown, and it is unclear whether they reflect the process of the COVID-19 infection. In addition, whether an acute cardiac injury is a primary infective phenomenon or secondary to lung disease is still unknown. While much remains unclear, we can still say with certainty that the detection of acute cardiac injury in hospitalized COVID-19 patients may help identify a subset of patients at greater risk of COVID-19 complications.
MYOCARDITIS:
COVID-19 patients have a broad spectrum of cardiovascular complications; among these complications, myocarditis is a potentially lethal complication [71]. Long-term experience tells us that acute heart injury and myocarditis are recognized complications of acute viral infections. The possible clinical symptoms of myocarditis range from fatigue, chest pain, and palpitations to life-threatening presentations such as cardiogenic shock or sudden cardiac death associated with ventricular arrhythmias [72]. Because of the expression ACE2, myocardial cells are a potential target of SARS-CoV-2. Myocarditis has been reported in a limited series in China, where 7% of deaths were attributed to myocardial damage with circulatory failure without a clear, definite diagnosis of myocarditis, and another 33% of patients died of circulatory failure caused by myocarditis [5]. Outside of published case reports, the exact incidence of myocardial involvement or myocarditis among COVID-19 patients is unknown. Viral myocarditis is a common cause of cardiac injury in patients with COVID-19; thus, it can indirectly indicate the existence of myocarditis. Myocarditis is known to result in focal or global myocardial inflammation, necrosis, and, eventually, ventricular dysfunction [68] (Figure 3). Myocyte necrosis and mononuclear cell infiltrates were found in COVID-19 patient autopsy specimens of the National Health Commission of the PRC [73], but there was no clear evidence of COVID-19 in the myocardium. A study found that, unlike findings in the lungs, there was no SARS-CoV-2 RNA in the myocardium of 5 patients who died due to COVID-19 [74].
Although the relevant evidence is insufficient, and the clinical symptoms are not very clear, we still need to pay attention to prevent the occurrence of myocarditis during the treatment of patients with new coronary disease.
CORONARY HEART DISEASE:
Preexisting cardiovascular diseases might be more susceptible to COVID-19 – induced heart injury, as approximately 30% of patients with cardiac injury in a study had a history of coronary heart disease [3]. Shi et al reported the importance of heart injury to COVID-19 mortality in 416 patients hospitalized with COVID-19, including 57 deaths. Of these patients, 10.6% had coronary heart disease [3]. Zhou et al drew a similar conclusion. They surveyed 191 patients with COVID-19. Among 91 (48%) patients with comorbidities, 15 (8%) had coronary heart disease [75].
In terms of mortality, compared with survivors, patients with comorbid coronary heart disease had a higher mortality rate (1% vs 24%) [75] (Figure 3), and patients with coronary heart disease were significantly more likely to die. Researchers have pointed out that the prognosis of patients with acute coronary syndrome (ACS) infected with SARS-CoV-2 is usually poor [7]. Myocardial ischemia and necrosis caused by ACS makes patients with COVID-19 more prone to the insufficient blood supply to the heart, which increases the risk of sudden deterioration of the patient’s condition [7].
ARRHYTHMIA:
COVID-19-related arrhythmia complications are relatively rare in existing reports. An early case series from China reported that 16.7% of 138 hospitalized COVID-19 patients developed arrhythmia [4]. Although the cause and type were not specified, this is still a high incidence that cannot be ignored. Interestingly, a report of the National Health Commission of China estimates that during the initial outbreak, some patients reported primarily CV symptoms, such as palpitations and chest tightness, rather than respiratory symptoms [7]. It was also found that patients admitted to the ICU had a higher incidence of arrhythmia (44.4% vs 6.9%, P<0.01) [4]. Previous historical epidemics, as SARS-CoV, MERS-CoV, and influenza, show us that outbreaks were usually associated with cardiovascular diseases like arrhythmia [76]. Yu et al confirmed that sinus tachycardia is the most common cardiovascular complication in SARS-CoV-infected patients, with a total incidence rate of 72%. This arrhythmia may be related to medication or autonomous regulation (Figure 1). At the same time, 14.9% of patients had obvious sinus bradycardia [77]. A study of 70 MERS-CoV patients found that 15.7% of those with complications had arrhythmia [78]. Given that SARS-CoV-2 has a similar structure to SARS-CoV and MERS-CoV, this novel virus might also have an interaction with arrhythmia. Furthermore, from the perspective of viral infectious diseases, Madjid et al has proved a link between arrhythmia and influenza [79]. This may be caused by increased oxygen demand of the patient and, potentially, acute coronary syndrome caused by influenza [79] (Figure 3). SARS-CoV-2 may have similar mechanisms.
HYPERTENSION:
Statistics of the first group of patients diagnosed with COVID-19 in Wuhan show that 32% had underlying diseases, including hypertension (15%) [2]. In another study, 46.4% of diagnosed patients had 1 or more coexisting medical conditions, among which hypertension (31.2%) was the most common coexisting condition [4]. In addition, patients with underlying diseases are more likely to need ICU care, including those with hypertension (21 [58.3%] vs 22 [21.6%]) [4], which means that their conditions are worse. This feature also is reflected in the case fatality rate; compared to survivors, patients with the comorbidity of hypertension have a higher mortality rate (23% vs 48%) [75]. Patients with hypertension more likely to be infected with SARS-CoV-2 and their underlying diseases may also have more severe symptoms after infection [7].
However, some studies also found that it is unclear whether hypertension is a risk factor for susceptibility to SARS-CoV-2 infection. After all, the available data show prevalence rates of COVID-19 in hypertensive patients are largely in line with rates of high blood pressure in the general population (~30%) [80]. Age is also an important factor. Older people have a higher prevalence of hypertension and diabetes, and older individuals have significantly worse outcomes, more severe course of COVID-19, and a higher mortality rate than the younger patients. Therefore, we cannot jump to conclusions about whether there is a causal relationship between hypertension and COVID-19.
However, in summary, while hypertension does appear to be clearly associated with COVID-19 infection, we can be sure that patients with preexisting cardiovascular conditions (hypertension in particular) had the highest morbidity (10.5%) following infection [81].
The putative relationship between hypertension and COVID-19 may relate to the role of ACE2. Besides the cellular receptor of SARS-CoV-2, ACE2 is a master regulator of the renin-angiotensin-aldosterone system (RAAS) [82]. Thus, both reduced or increased function of ACE2 can induce systemic and pulmonary hypertension, heart failure, myocardial infarction, and diabetic cardiovascular complications [83]. Angiotensin-converting enzyme inhibitors (ACEI) and angiotensin II receptor blockers (ARBs) are both RAAS inhibitors. Through their corresponding substrates, SARS-CoV-2 binding is unlikely to have any relationship with ACE or ACEI [82], but in light of the route of SARS-CoV-2 infection, the role of ACE2 and ARB should not be underestimated. There is some evidence that ARBs can modify ACE2 expression, primarily by upregulation in cardiac tissue and renal vasculature [84,85]. Some studies have found ARBs are commonly used to protect hypertension and maintain insulin sensitivity [86,87]. Consequently, the increased expression of ACE2 would facilitate infection with COVID-19 [88]. Moreover, it also speeds the development of COVID-19 and increases the mortality rate [88] (Figure 3). Thus, in theory, this treatment may increase the risk of COVID-19 infection. However, there is insufficient evidence that ARBs or ACEIs can facilitate SARS-CoV-2 entry and cause COVID-19.
The role of the immune system also cannot be ignored. Poor control of blood pressure may contribute to further dysregulation of the immune system. For example, monocytes can be activated by the vascular endothelium during hypertension, which is related to the massive release of cytokines [89], which is a situation providing a possible link to COVID-19. CD8+ T cell dysfunction is also observed in patients with hypertension. Such pathological changes make it unable to play an efficient role in normal immune defense function, and finally, contribute to pathological over-production of cytokines [90].
Overall, it is essential to ensure blood pressure control and blood glucose balance, and attention must be paid to carefully monitoring for CV and other complications during COVID-19 infection.
Treatment
ARBS FOR HYPERTENSION AND DIABETES:
Hypertension and diabetes, as common complications in patients with COVID-19, are often treated with ARBs [86,87]. However, this treatment is a double-edged sword in the progression of the COVID-19. ARBs can modify ACE2 expression, primarily upregulating in the cardiac tissue and the renal vasculature [84,85]. ACE2 acts as a receptor for SARS-CoV-2 binding. Thus, use of ARBs may accelerate COVID-19 while treating hypertension, but controlling blood pressure and blood glucose is also crucial for patients with COVID-19. Attention must be paid to monitoring for CV and other complications during COVID-19 infection when using ARBs. Alternative treatments can also be used: antihypertensive calcium channel blockers have little effect on the expression and activity of ACE2; therefore, they could be a suitable alternative treatment in these patients [88].
CHLOROQUINE AND ARRHYTHMIAS:
During the epidemic of SARS-CoV-2, both chloroquine and hydroxychloroquine have shown certain curative effects in controlling the severity of illness of COVID-19 patients, manifested as reduced pneumonia severity, hospitalization length, and viral shedding [60]. However, these 2 drugs have been reported to be associated with cardiac toxicity, even at therapeutic doses, manifested as abnormal electrocardiographic changes (eg, QT prolongation [91], T-wave inversion/depression, and widened QRS complex [92]) and increased risk of TdP, and have been listed as agents with myocardial toxicity by the American Heart Association [93]. Administration of chloroquine and hydroxychloroquine combined with azithromycin (another agent associated with malignant arrhythmias and widely used in SARS-CoV-2 infection [94]) or lopinavir/ritonavir can elevate plasma levels of chloroquine and further increase the risk of acute arrhythmias [95]. Therefore, combined therapy of chloroquine/hydroxychloroquine and QTc-prolonging medications or lopinavir/ritonavir should be used carefully. Due to the lack of effective medications to reduce the risks of arrhythmias caused by chloroquine and hydroxychloroquine, when using these 2 agents in COVID-19 patients, after weighing the advantages and disadvantages, clinical decision-makers should closely monitor for changes in ECG and related biochemical indexes in patients in case of cardiovascular complications.
CYTOKINE DETECTION:
Inflammatory factors are an important mechanism in the development of the COVID-19, and can also cause many complications, such as cardiovascular diseases. Among inflammatory factors, IL-6 has a dual effect on the myocardium. IL-6 participates in the pathogenesis of cardiac dysfunctions. With IL-6 levels >1.5 ng/L, the risk of cardiovascular death starts to increase to an almost 4-fold difference among those with the highest values [42]. However, some studies have found that IL-6 also has a protective effect on the heart. IL-6 plays an important role in myocardial remodeling through activating TGF-β1 signaling to reprogram the processes of proliferation and/or fibrosis in fibroblasts [44]. IL-6 can also regulate inflammasome activation and consequently suppress the over-production of nitric oxide, which contributes to lethal oxidative stress [45]. The different effects may be due to the content of IL-6. Based on the duality of inflammatory factors in the course of the COVID-19, we should monitor them closely.
Conclusions
COVID-19 infection can affect the cardiovascular system. Many clinical data and reports that have been published show that cardiovascular diseases such as myocarditis, hypertension, and arrhythmia are closely related to COVID-19, and direct cell damage through ACE2, stress, hypoxia, inflammatory factors, and drug use are all possible mechanisms. While it is unclear whether the cardiovascular disease is secondary or primary, it is clear that COVID-19 patients with cardiovascular diseases often have higher mortality and worse prognosis. Attention should focus on both the mechanism and clinical data to protect the cardiovascular system when treating patients with COVID-19.
Figures
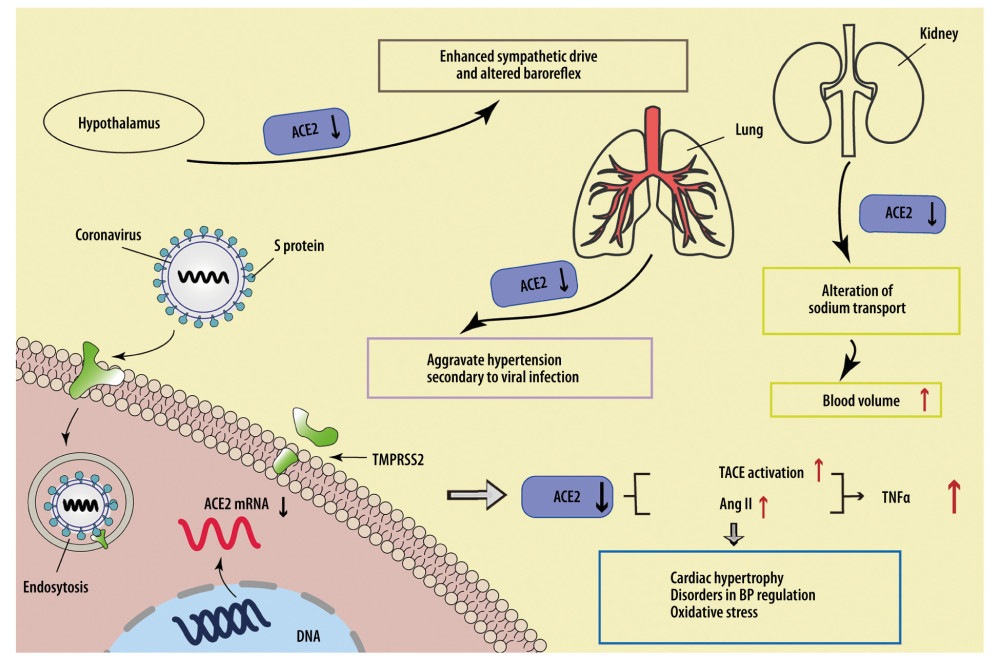 Figure 1. SARS-CoV-2 downregulates ACE2 to cause cardiovascular injuries. SARS-CoV-2 downregulates ACE2 by suppressing transcription of the ACE2 gene, cutting the extracellular segment of ACE2, and increasing internalization with ACE2, leading to increased Ang II expression and TACE activation, thus elevating the expression level of TNF-α and the risk of cardiovascular adverse events. The downregulation of ACE2 occurring in the heart, lung, hypothalamus, and kidney can all increase the load on the cardiovascular system.
Figure 1. SARS-CoV-2 downregulates ACE2 to cause cardiovascular injuries. SARS-CoV-2 downregulates ACE2 by suppressing transcription of the ACE2 gene, cutting the extracellular segment of ACE2, and increasing internalization with ACE2, leading to increased Ang II expression and TACE activation, thus elevating the expression level of TNF-α and the risk of cardiovascular adverse events. The downregulation of ACE2 occurring in the heart, lung, hypothalamus, and kidney can all increase the load on the cardiovascular system. 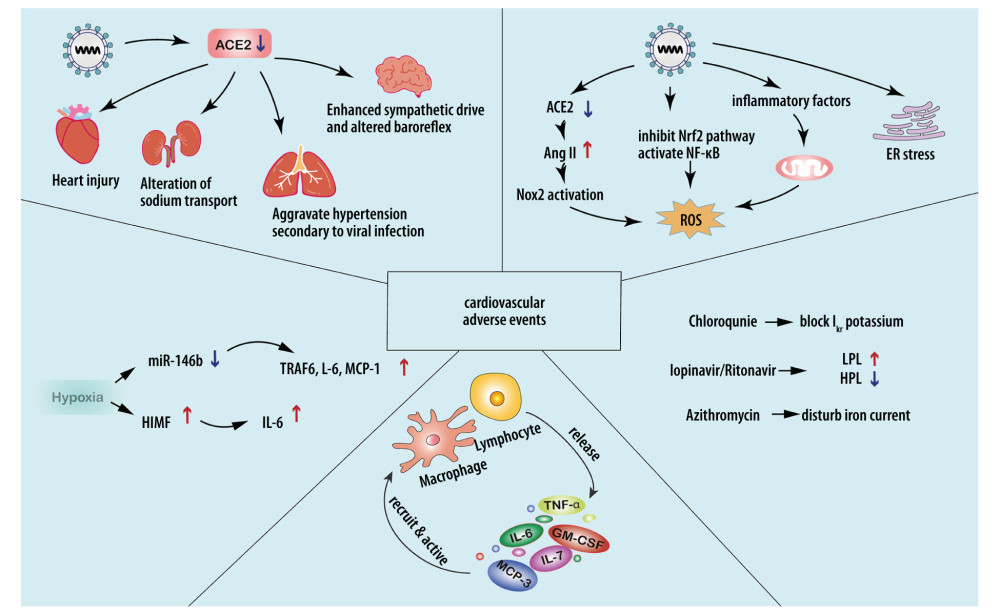 Figure 2. Possible mechanisms for cardiovascular injuries after SARS-CoV-2 infection. a) Stress: Downregulation of ACE2 could lead to excessive Nox2 activation, inducing ROS accumulation. High levels of TNF-α and IL-6 can increase mitochondrial stress, leading to higher levels of oxidative stress. ER stress can destabilize atherosclerotic plaque through increasing NF-κB-induced GM-CSF. b) Hypoxia: SARS-CoV-2-related hypoxemia can affect the miR-146b-TRAF6-IL-6/CCL2 (MCP-1) axis and induce expression of HIMF, promoting the production of proinflammatory cytokines. c) Accumulation of proinflammatory cytokines contributes to cardiovascular complications. d) Drug-related injuries: Drugs administered in treating SARS-CoV-2 can cause cardiovascular complications. Chloroquine, lopinavir/ritonavir, and azithromycin can affect the normal rhythm of the heart. Lopinavir/ritonavir can also increase LDL and suppress HDL, destabilizing atherosclerotic plaque. Pegylated interferon-α can lead to the upregulation of TNF-α.
Figure 2. Possible mechanisms for cardiovascular injuries after SARS-CoV-2 infection. a) Stress: Downregulation of ACE2 could lead to excessive Nox2 activation, inducing ROS accumulation. High levels of TNF-α and IL-6 can increase mitochondrial stress, leading to higher levels of oxidative stress. ER stress can destabilize atherosclerotic plaque through increasing NF-κB-induced GM-CSF. b) Hypoxia: SARS-CoV-2-related hypoxemia can affect the miR-146b-TRAF6-IL-6/CCL2 (MCP-1) axis and induce expression of HIMF, promoting the production of proinflammatory cytokines. c) Accumulation of proinflammatory cytokines contributes to cardiovascular complications. d) Drug-related injuries: Drugs administered in treating SARS-CoV-2 can cause cardiovascular complications. Chloroquine, lopinavir/ritonavir, and azithromycin can affect the normal rhythm of the heart. Lopinavir/ritonavir can also increase LDL and suppress HDL, destabilizing atherosclerotic plaque. Pegylated interferon-α can lead to the upregulation of TNF-α. 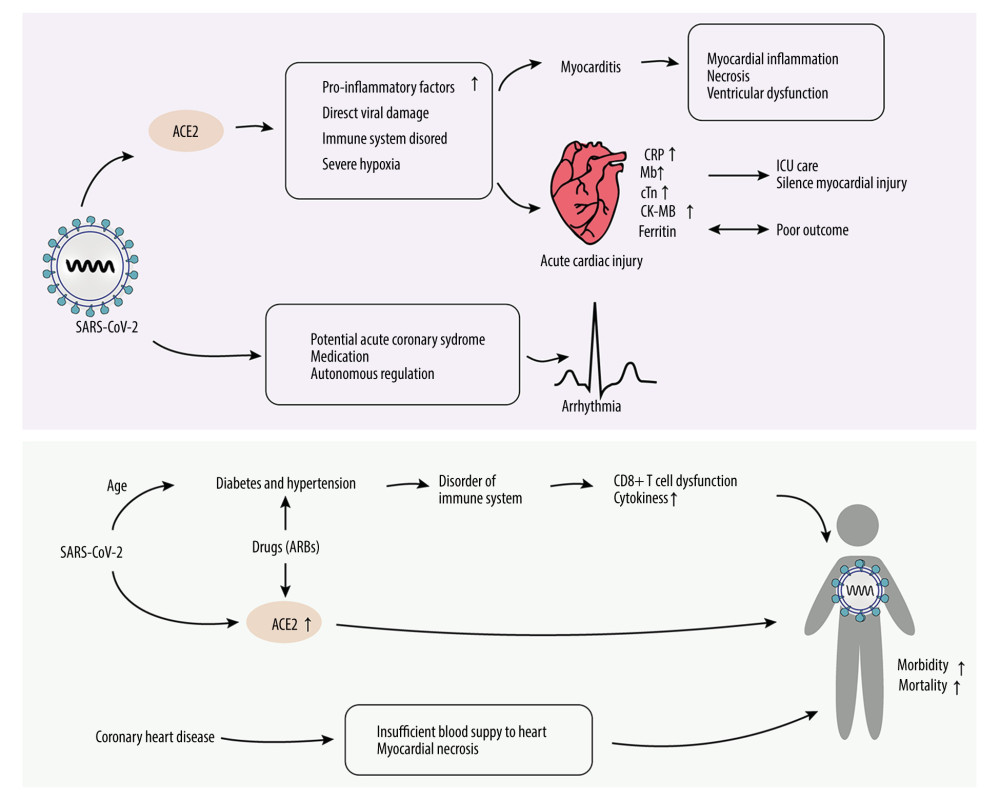 Figure 3. Injury mechanism of SARS-CoV-2 on cardiovascular system. After SARS-CoV-2 binds to the ACE2 receptor, it causes cardiovascular diseases through a variety of ways, such as acute cardiac injury, myocarditis, arrhythmia, coronary heart diseases, diabetes, and hypertension, and further results in higher morbidity. Among all myocardial injury markers, ferritin is most associated with prognosis. Mb changes are related to ICU care and silent myocardial injury. Hypertension and diabetes can affect the immune system and its drug therapy. ARBs can upregulate the expression of ACE2 and aggravate the process of COVID-19. Mb – creatine kinase-MB; ARBs – angiotensin II receptor blockers.
Figure 3. Injury mechanism of SARS-CoV-2 on cardiovascular system. After SARS-CoV-2 binds to the ACE2 receptor, it causes cardiovascular diseases through a variety of ways, such as acute cardiac injury, myocarditis, arrhythmia, coronary heart diseases, diabetes, and hypertension, and further results in higher morbidity. Among all myocardial injury markers, ferritin is most associated with prognosis. Mb changes are related to ICU care and silent myocardial injury. Hypertension and diabetes can affect the immune system and its drug therapy. ARBs can upregulate the expression of ACE2 and aggravate the process of COVID-19. Mb – creatine kinase-MB; ARBs – angiotensin II receptor blockers. References
1. World Health Organization: WHO Coronavirus Disease (COVID-19) Dashboard, 2020 [cited 2020, 8/31]. https://covid19.who.int/
2. Huang C, Wang Y, Li X, Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China: Lancet, 2020; 395(10223); 497-506
3. Shi S, Qin M, Shen B, Association of cardiac injury with mortality in hospitalized patients with COVID-19 in Wuhan, China: JAMA Cardiol, 2020; 5(7); 802-10
4. Wang D, Hu B, Hu C, Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus-infected pneumonia in Wuhan, China: JAMA, 2020; 323(11); 1061-69
5. Ruan Q, Yang K, Wang W, Clinical predictors of mortality due to COVID-19 based on an analysis of data of 150 patients from Wuhan, China: Intensive Care Med, 2020; 46(5); 846-48
6. Wu Z, McGoogan JM, Characteristics of and important lessons from the coronavirus disease 2019 (COVID-19) outbreak in China: Summary of a report of 72 314 cases from the Chinese center for disease control and prevention: JAMA, 2020; 323(13); 1239-42
7. Zheng YY, Ma YT, Zhang JY, COVID-19 and the cardiovascular system: Nat Rev Cardiol, 2020; 17(5); 259-60
8. Zhu H, Rhee JW, Cheng P, Cardiovascular complications in patients with COVID-19: Consequences of viral toxicities and host immune response: Curr Cardiol Rep, 2020; 22(5); 32
9. Turner AJ, Hiscox JA, Hooper NM, ACE2: from vasopeptidase to SARS virus receptor: Trends Pharmacol Sci, 2004; 25(6); 291-94
10. Sharma A, Garcia G, Arumugaswami V, Human iPSC-derived cardiomyocytes are susceptible to SARS-CoV-2 infection: bioRxiv, 2020; 2020; 051912
11. Chen L, Li X, Chen M, The ACE2 expression in human heart indicates new potential mechanism of heart injury among patients infected with SARS-CoV-2: Cardiovasc Res, 2020; 116(6); 1097-100
12. Haga S, Yamamoto N, Nakai-Murakami C, Modulation of TNF-alpha-converting enzyme by the spike protein of SARS-CoV and ACE2 induces TNF-alpha production and facilitates viral entry: Proc Natl Acad Sci USA, 2008; 105(22); 7809-14
13. Hoffmann M, Kleine-Weber H, Schroeder S, SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor: Cell, 2020; 181(2); 271-80.e8
14. Verdecchia P, Cavallini C, Spanevello A, The pivotal link between ACE2 deficiency and SARS-CoV-2 infection: Eur J Intern Med, 2020; 76; 14-20
15. Kuba K, Imai Y, Rao S, A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury: Nat Med, 2005; 11(8); 875-79
16. Wu C-H, Mohammadmoradi S, Chen JZ, Renin-angiotensin system and cardiovascular functions: Arterioscler Thromb Vasc Biol, 2018; 38(7); e108-e16
17. Miller AJ, Arnold AC, The renin-angiotensin system in cardiovascular autonomic control: Recent developments and clinical implications: Clin Auton Res, 2019; 29(2); 231-43
18. Recinos A, LeJeune WS, Sun H, Angiotensin II induces IL-6 expression and the Jak-STAT3 pathway in aortic adventitia of LDL receptor-deficient mice: Atherosclerosis, 2007; 194(1); 125-33
19. Yamamoto S, Yancey PG, Zuo Y, Macrophage polarization by angiotensin II-type 1 receptor aggravates renal injury-acceleration of atherosclerosis: Arterioscler Thromb Vasc Biol, 2011; 31(12); 2856-64
20. Long B, Brady WJ, Koyfman A, Cardiovascular complications in COVID-19: Am J Emerg Med, 2020; 38(7); 1504-7
21. Wichmann D, Sperhake JP, Lütgehetmann M, Autopsy findings and venous thromboembolism in patients with COVID-19: A prospective cohort study: Ann Intern Med, 2020; 173(4); 268-77
22. Puelles VG, Lütgehetmann M, Lindenmeyer MT, Multiorgan and renal tropism of SARS-CoV-2: N Engl J Med, 2020; 383(6); 590-92
23. Chai X, Hu L, Zhang Y, Specific ACE2 expression in cholangiocytes may cause liver damage after 2019-nCoV infection: bioRxiv, 2020; 2020; 931766
24. Alenina N, Bader M, ACE2 in brain physiology and pathophysiology: Evidence from transgenic animal models: Neurochem Res, 2019; 44(6); 1323-29
25. South AM, Diz DI, Chappell MC, COVID-19, ACE2, and the cardiovascular consequences: Am J Physiol Heart Circ Physiol, 2020; 318(5); H1084-90
26. Guo T, Fan Y, Chen M, Cardiovascular implications of fatal outcomes of patients with coronavirus disease 2019 (COVID-19): JAMA Cardiol, 2020; 5(7); 1-8
27. Khan Z, Shen XZ, Bernstein EA, Angiotensin-converting enzyme enhances the oxidative response and bactericidal activity of neutrophils: Blood, 2017; 130(3); 328-39
28. Cangemi R, Calvieri C, Bucci T, Is NOX2 upregulation implicated in myocardial injury in patients with pneumonia?: Antioxid Redox Signal, 2014; 20(18); 2949-54
29. Cangemi R, Celestini A, Del Ben M, Role of platelets in NOX2 activation mediated by TNFα in heart failure: Intern Emerg Med, 2014; 9(2); 179-85
30. Komaravelli N, Casola A, Respiratory viral infections and subversion of cellular antioxidant defenses: J Pharmacogenomics Pharmacoproteomics, 2014; 5(4); 1000141
31. Shao H, Lan D, Duan Z, Upregulation of mitochondrial gene expression in PBMC from convalescent SARS patients: J Clin Immunol, 2006; 26(6); 546-54
32. Green DR, Galluzzi L, Kroemer G, Mitochondria and the autophagy-inflammation-cell death axis in organismal aging: Science, 2011; 333(6046); 1109-12
33. Saleh J, Peyssonnaux C, Singh KK, Mitochondria and microbiota dysfunction in COVID-19 pathogenesis: Mitochondrion, 2020; 54; 1-7
34. Chen AC, Burr L, McGuckin MA, Oxidative and endoplasmic reticulum stress in respiratory disease: Clin Transl Immunol, 2018; 7(6); e1019
35. Katzen J, Wagner BD, Venosa A, An SFTPC BRICHOS mutant links epithelial ER stress and spontaneous lung fibrosis: JCI Insight, 2019; 4(6); e126125
36. Omidkhoda N, Wallace Hayes A, Reiter RJ, The role of MicroRNAs on endoplasmic reticulum stress in myocardial ischemia and cardiac hypertrophy: Pharmacol Res, 2019; 150; 104516
37. Ying R, Li SW, Chen JY, Endoplasmic reticulum stress in perivascular adipose tissue promotes destabilization of atherosclerotic plaque by regulating GM-CSF paracrine: J Transl Med, 2018; 16(1); 105
38. Brito-Azevedo A, Pinto EC, de Cata Preta Corrêa GA, SARS-CoV-2 infection causes pulmonary shunt by vasodilatation: J Med Virol, 2021; 93(1); 573-75
39. Chouvarine P, Legchenko E, Geldner J, Hypoxia drives cardiac miRNAs and inflammation in the right and left ventricle: J Mol Med, 2019; 97(10); 1427-38
40. Angelini DJ, Su Q, Yamaji-Kegan K, Hypoxia-induced mitogenic factor (HIMF/FIZZ1/RELMalpha) induces the vascular and hemodynamic changes of pulmonary hypertension: Am J Physiol Lung Cell Mol Physiol, 2009; 296(4); L582-93
41. Kumar S, Wang G, Zheng N, HIMF (hypoxia-induced mitogenic factor)-IL (Interleukin)-6 signaling mediates cardiomyocyte-fibroblast crosstalk to promote cardiac hypertrophy and fibrosis: Hypertension, 2019; 73(5); 1058-70
42. Held C, White HD, Stewart RAH, Inflammatory biomarkers Interleukin-6 and C-reactive protein and outcomes in stable coronary heart disease: Experiences from the STABILITY (stabilization of atherosclerotic plaque by initiation of darapladib therapy) trial: J Am Heart Assoc, 2017; 6(10); e005077
43. Jong WM, Ten Cate H, Linnenbank AC, Reduced acute myocardial ischemia-reperfusion injury in IL-6-deficient mice employing a closed-chest model: Inflamm Res, 2016; 65(6); 489-99
44. Jing R, Long TY, Pan W, IL-6 knockout ameliorates myocardial remodeling after myocardial infarction by regulating activation of M2 macrophages and fibroblast cells: Eur Rev Med Pharmacol Sci, 2019; 23(14); 6283-91
45. Sanmarco LM, Ponce NE, Visconti LM, IL-6 promotes M2 macrophage polarization by modulating purinergic signaling and regulates the lethal release of nitric oxide during Trypanosoma cruzi infection: Biochim Biophys Acta Mol Basis Dis, 2017; 1863(4); 857-69
46. Xu Y, Zhang Y, Ye J, IL-6: A potential role in cardiac metabolic homeostasis: Int J Mol Sci, 2018; 19(9); 2474
47. D’Souza K, Nzirorera C, Kienesberger PC, Lipid metabolism and signaling in cardiac lipotoxicity: Biochim Biophys Acta, 2016; 1861(10); 1513-24
48. Wang J, Jiang M, Chen X, Cytokine storm and leukocyte changes in mild versus severe SARS-CoV-2 infection: Review of 3939 COVID-19 patients in China and emerging pathogenesis and therapy concepts: J Leukoc Biol, 2020; 108(1); 17-41
49. Garcia B, Cousin N, Bourel C, Prone positioning under VV-ECMO in SARS-CoV-2-induced acute respiratory distress syndrome: Crit Care, 2020; 24(1); 428
50. Hartman ME, Hernandez RA, Patel K, COVID-19 respiratory failure: Targeting inflammation on VV-ECMO support: ASAIO J, 2020; 66(6); 603-6
51. Kowalewski M, Fina D, Słomka A, COVID-19 and ECMO: The interplay between coagulation and inflammation-a narrative review: Crit Care, 2020; 24(1); 205
52. Gralinski LE, Sheahan TP, Morrison TE, Complement activation contributes to severe acute respiratory syndrome coronavirus pathogenesis: mBio, 2018; 9(5); e01753-18
53. Huang C, Wang Y, Li X, Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China: Lancet, 2020; 395(10223); 497-506
54. Savarino A, Boelaert JR, Cassone A, Effects of chloroquine on viral infections: An old drug against today’s diseases?: Lancet Infect Dis, 2003; 3(11); 722-27
55. Principi N, Esposito S, Chloroquine or hydroxychloroquine for prophylaxis of COVID-19: Lancet Infect Dis, 2020; 20(10); 1118
56. Bauman JL, Tisdale JE, Chloroquine and hydroxychloroquine in the era of SARS-CoV2: Caution on their cardiac toxicity: Pharmacotherapy, 2020; 40(5); 387-88
57. Cao B, Wang Y, Wen D, A trial of lopinavir-ritonavir in adults hospitalized with severe COVID-19: N Engl J Med, 2020; 382(19); 1787-99
58. Limsreng S, Marcy O, Ly S, Dyslipidemias and elevated cardiovascular risk on lopinavir-based antiretroviral therapy in Cambodia: PLoS One, 2016; 11(8); e0160306
59. Mulangu S, Dodd LE, Davey RT, A randomized, controlled trial of Ebola virus disease therapeutics: N Engl J Med, 2019; 381(24); 2293-303
60. Gautret P, Lagier JC, Parola P, Hydroxychloroquine and azithromycin as a treatment of COVID-19: Results of an open-label non-randomized clinical trial: Int J Antimicrob Agents, 2020; 56(1); 105949
61. Yang Z, Prinsen JK, Bersell KR, Azithromycin causes a novel proarrhythmic syndrome: Circ Arrhythm Electrophysiology, 2017; 10(4); e003560
62. , In brief: FDA azithromycin warning: Med Lett Drugs Ther, 2013; 55(1413); 28
63. Aggarwal G, Henry BM, Aggarwal S, Cardiovascular safety of potential drugs for the treatment of coronavirus disease 2019: Am J Cardiol, 2020; 128; 147-50
64. Teragawa H, Hondo T, Amano H, Adverse effects of interferon on the cardiovascular system in patients with chronic hepatitis C: Jpn Heart J, 1996; 37(6); 905-15
65. Alhogbani T, Acute myocarditis associated with novel Middle east respiratory syndrome coronavirus: Ann Saudi Med, 2016; 36(1); 78-80
66. Alexander LK, Small JD, Edwards S, An experimental model for dilated cardiomyopathy after rabbit coronavirus infection: J Infect Dis, 1992; 166(5); 978-85
67. Riski H, Hovi T, Frick MH, Carditis associated with coronavirus infection: Lancet, 1980; 2(8185); 100-1
68. Inciardi RM, Lupi L, Zaccone G, Cardiac involvement in a patient with coronavirus disease 2019 (COVID-19): JAMA Cardiol, 2020; 5(7); 1-6
69. Guzik TJ, Mohiddin SA, Dimarco A, COVID-19 and the cardiovascular system: Implications for risk assessment, diagnosis, and treatment options: Cardiovasc Res, 2020; 116(10); 1666-87
70. De Lorenzo A, Kasal DA, Tura BR, Acute cardiac injury in patients with COVID-19: Am J Cardiovasc Dis, 2020; 10(2); 28-33
71. Pirzada A, Mokhtar AT, Moeller AD, COVID-19 and myocarditis: What do we know so far?: CJC Open, 2020; 2(4); 278-85
72. Caforio AL, Pankuweit S, Arbustini E, Current state of knowledge on aetiology, diagnosis, management, and therapy of myocarditis: A position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases: Eur Heart J, 2013; 34(33); 2636-48
73. PRC tNHCot: Chinese Clinical Guidance for COVID-19 Pneumonia Diagnosis and Treatment, 2020 [cited 2020 03, 16]. http://kjfy.meetingchina.org/msite/news/show/cn/3337.html
74. Imazio M, Klingel K, Kindermann I, COVID-19 pandemic and troponin: Indirect myocardial injury, myocardial inflammation or myocarditis?: Heart, 2020; 106(15); 1127-31
75. Zhou F, Yu T, Du R, Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study: Lancet, 2020; 395(10229); 1054-62
76. Kochi AN, Tagliari AP, Forleo GB, Cardiac and arrhythmic complications in patients with COVID-19: J Cardiovasc Electrophysiol, 2020; 31(5); 1003-8
77. Yu CM, Wong RS, Wu EB, Cardiovascular complications of severe acute respiratory syndrome: Postgrad Med J, 2006; 82(964); 140-44
78. Saad M, Omrani AS, Baig K, Clinical aspects and outcomes of 70 patients with Middle East respiratory syndrome coronavirus infection: A single-center experience in Saudi Arabia: Int J Infect Dis, 2014; 29; 301-6
79. Madjid M, Connolly AT, Nabutovsky Y, Effect of high influenza activity on risk of ventricular arrhythmias requiring therapy in patients with implantable cardiac defibrillators and cardiac resynchronization therapy defibrillators: Am J Cardiol, 2019; 124(1); 44-50
80. Beaney T, Burrell LM, Castillo RR, May Measurement Month 2018: A pragmatic global screening campaign to raise awareness of blood pressure by the International Society of Hypertension: Eur Heart J, 2019; 40(25); 2006-17
81. Driggin E, Madhavan MV, Bikdeli B, Cardiovascular considerations for patients, health care workers, and health systems during the COVID-19 pandemic: J Am Coll Cardiol, 2020; 75(18); 2352-71
82. Tadic M, Cuspidi C, Mancia G, COVID-19, hypertension and cardiovascular diseases: Should we change the therapy?: Pharmacol Res, 2020; 158; 104906
83. Patel VB, Zhong JC, Grant MB, Role of the ACE2/Angiotensin 1–7 axis of the renin-angiotensin system in heart failure: Circ Res, 2016; 118(8); 1313-26
84. Wang X, Ye Y, Gong H, The effects of different angiotensin II type 1 receptor blockers on the regulation of the ACE-AngII-AT1 and ACE2-Ang(1–7)-Mas axes in pressure overload-induced cardiac remodeling in male mice: J Mol Cell Cardiol, 2016; 97; 180-90
85. Soler MJ, Ye M, Wysocki J, Localization of ACE2 in the renal vasculature: Amplification by angiotensin II type 1 receptor blockade using telmisartan: Am J Physiol Renal Physiol, 2009; 296(2); F398-405
86. Wan Y, Shang J, Graham R, Receptor recognition by the novel coronavirus from Wuhan: An analysis based on decade-long structural studies of SARS coronavirus: J Virol, 2020; 94(7); e00127-20
87. Li XC, Zhang J, Zhuo JL, The vasoprotective axes of the renin-angiotensin system: Physiological relevance and therapeutic implications in cardiovascular, hypertensive and kidney diseases: Pharmacol Res, 2017; 125(Pt A); 21-38
88. Fang L, Karakiulakis G, Roth M, Are patients with hypertension and diabetes mellitus at increased risk for COVID-19 infection?: Lancet Respir Med, 2020; 8(4); e21
89. Loperena R, Van Beusecum JP, Itani HA, Hypertension and increased endothelial mechanical stretch promote monocyte differentiation and activation: Roles of STAT3, interleukin 6 and hydrogen peroxide: Cardiovasc Res, 2018; 114(11); 1547-63
90. Youn JC, Yu HT, Lim BJ, Immunosenescent CD8+ T cells and C-X-C chemokine receptor type 3 chemokines are increased in human hypertension: Hypertension, 2013; 62(1); 126-33
91. Wozniacka A, Cygankiewicz I, Chudzik M, The cardiac safety of chloroquine phosphate treatment in patients with systemic lupus erythematosus: The influence on arrhythmia, heart rate variability and repolarization parameters: Lupus, 2006; 15(8); 521-25
92. Burrell ZL, Martinez AC, Chloroquine and hydroxychloroquine in the treatment of cardiac arrhythmias: N Engl J Med, 1958; 258(16); 798-800
93. Page RL, O’Bryant CL, Cheng D, Drugs that may cause or exacerbate heart failure: A scientific statement from the American Heart Association: Circulation, 2016; 134(6); e32-69
94. Mercuro NJ, Yen CF, Shim DJ, Risk of QT interval prolongation associated with use of hydroxychloroquine with or without concomitant azithromycin among hospitalized patients testing positive for coronavirus disease 2019 (COVID-19): JAMA Cardiol, 2020; 5(9); 1036-41
95. Pastick KA, Okafor EC, Wang F, Review: Hydroxychloroquine and chloroquine for treatment of SARS-CoV-2 (COVID-19): Open Forum Infect Dis, 2020; 7(4); ofaa130
Figures
Figure 1. SARS-CoV-2 downregulates ACE2 to cause cardiovascular injuries. SARS-CoV-2 downregulates ACE2 by suppressing transcription of the ACE2 gene, cutting the extracellular segment of ACE2, and increasing internalization with ACE2, leading to increased Ang II expression and TACE activation, thus elevating the expression level of TNF-α and the risk of cardiovascular adverse events. The downregulation of ACE2 occurring in the heart, lung, hypothalamus, and kidney can all increase the load on the cardiovascular system.Figure 2. Possible mechanisms for cardiovascular injuries after SARS-CoV-2 infection. a) Stress: Downregulation of ACE2 could lead to excessive Nox2 activation, inducing ROS accumulation. High levels of TNF-α and IL-6 can increase mitochondrial stress, leading to higher levels of oxidative stress. ER stress can destabilize atherosclerotic plaque through increasing NF-κB-induced GM-CSF. b) Hypoxia: SARS-CoV-2-related hypoxemia can affect the miR-146b-TRAF6-IL-6/CCL2 (MCP-1) axis and induce expression of HIMF, promoting the production of proinflammatory cytokines. c) Accumulation of proinflammatory cytokines contributes to cardiovascular complications. d) Drug-related injuries: Drugs administered in treating SARS-CoV-2 can cause cardiovascular complications. Chloroquine, lopinavir/ritonavir, and azithromycin can affect the normal rhythm of the heart. Lopinavir/ritonavir can also increase LDL and suppress HDL, destabilizing atherosclerotic plaque. Pegylated interferon-α can lead to the upregulation of TNF-α.Figure 3. Injury mechanism of SARS-CoV-2 on cardiovascular system. After SARS-CoV-2 binds to the ACE2 receptor, it causes cardiovascular diseases through a variety of ways, such as acute cardiac injury, myocarditis, arrhythmia, coronary heart diseases, diabetes, and hypertension, and further results in higher morbidity. Among all myocardial injury markers, ferritin is most associated with prognosis. Mb changes are related to ICU care and silent myocardial injury. Hypertension and diabetes can affect the immune system and its drug therapy. ARBs can upregulate the expression of ACE2 and aggravate the process of COVID-19. Mb – creatine kinase-MB; ARBs – angiotensin II receptor blockers. In Press
Clinical Research
Effects of Single-Bout Endurance Exercise Intensity on Peripheral Neurotrophic Factors in Patients With Isc...Med Sci Monit In Press; DOI: 10.12659/MSM.952089
Review article
Anisodus tanguticus in Cancer Research: A Review of Traditional Use, Phytochemistry, Extraction Methods, an...Med Sci Monit In Press; DOI: 10.12659/MSM.952999
Clinical Research
Nasal Mucociliary Clearance and Its Relationship With Disease Severity in Patients With Multiple SclerosisMed Sci Monit In Press; DOI: 10.12659/MSM.952850
Clinical Research
Modified Thoracoabdominal Nerves Block Through the Perichondrial Approach vs Subcostal Transversus Abdomini...Med Sci Monit In Press; DOI: 10.12659/MSM.953976
Most Viewed Current Articles
17 Jan 2024 : Review article 14,176,570
Vaccination Guidelines for Pregnant Women: Addressing COVID-19 and the Omicron VariantDOI :10.12659/MSM.942799
Med Sci Monit 2024; 30:e942799
13 Nov 2021 : Clinical Research 3,762,188
Acceptance of COVID-19 Vaccination and Its Associated Factors Among Cancer Patients Attending the Oncology ...DOI :10.12659/MSM.932788
Med Sci Monit 2021; 27:e932788
14 Dec 2022 : Clinical Research 2,466,310
Prevalence and Variability of Allergen-Specific Immunoglobulin E in Patients with Elevated Tryptase LevelsDOI :10.12659/MSM.937990
Med Sci Monit 2022; 28:e937990
16 May 2023 : Clinical Research 708,927
Electrophysiological Testing for an Auditory Processing Disorder and Reading Performance in 54 School Stude...DOI :10.12659/MSM.940387
Med Sci Monit 2023; 29:e940387