11 November 2022: Review Articles
Autophagy and Its Association with Genetic Mutations in Parkinson Disease
Nour S. Erekat1EF*DOI: 10.12659/MSM.938519
Med Sci Monit 2022; 28:e938519






Abstract
ABSTRACT: Parkinson disease is the second most common neurodegenerative disorder, affecting 0.1-0.2% of the general population. It is a progressive debilitating disorder caused by degeneration of dopaminergic neurons in the substantia nigra pars compacta. It is characterized by motor and non-motor symptoms. Parkinson disease can be caused by mutations in genes that encode proteins involved in the autophagic process, resulting in impaired autophagy. Indeed, autophagy has been implicated in the pathogenesis of Parkinson disease, particularly because its impairment causes the buildup of proteins. Thus, this review aims to provide an overview of Parkinson disease-related genetic mutations and their association with autophagy impairment in Parkinson disease, which can be helpful in improving the understanding of the pathogenesis of Parkinson disease, illustrating the potential therapeutic implications of agents that can enhance autophagy in Parkinson disease. Additionally, we will highlight the essential need for the development of highly sensitive and specific assays for gene-based diagnostic biomarkers. Finally, we will provide an overview on the potential gene-based therapeutic approaches for Parkinson disease, which have been most advanced and are associated with the most common targets being alpha-synuclein (SNCA), leucine-rich repeat kinase-2 (LRRK2), and glucocerebrosidase (GBA).
Keywords: alpha-Synuclein, Autophagy, Mutation, Parkinson Disease, Humans, Glucosylceramidase, dopaminergic neurons
Background
Parkinson disease is the second most common neurodegenerative disorder, affecting 0.1–0.2% of the general population [1,2]. Its prevalence increases with age, affecting 1% and 4% of individuals who are older than 60 years and 80 years, respectively [1,3].
Parkinson disease is a progressive disorder caused by degeneration of dopaminergic neurons in the substantia nigra pars compacta [4–8]. It is characterized by motor and non-motor symptoms [1]. Non-motor symptoms often precede the motor symptoms [1]. Chief motor symptoms characteristic of Parkinson disease are at-rest tremor, bradykinesia, rigidity, and postural instability [1,9]. Thus, Parkinson disease is a debilitating disorder that can have a serious impact on the quality of life [1].
Parkinson disease can be either familial or sporadic [10]. Familial Parkinson disease cases are due to autosomal dominant and autosomal recessive mutations [10]. Parkinson disease is characterized by the presence of Lewy bodies, which are aggregated protein inclusions that contain α-synuclein as their main constituent [6].
Autophagy is a form of programmed cell death that is required for the turnover of long-lived organelles and elimination of impaired proteins [11]. Impaired autophagy has been shown to be involved in the pathogenesis of Parkinson disease, since it can lead to disrupted elimination of α-synuclein, resulting in its build up and subsequent misfolding [12,13]. Consequently, misfolded α-synuclein can participate in the formation of Lewy bodies, which are considered as the pathological hallmark in Parkinson disease [14,15].
Thus, this article aims to review the process of autophagy and its role in Parkinson disease, illustrating its association with the various genetic mutations of Parkinson disease, and highlighting its potential therapeutic implications in Parkinson disease. It also aims to highlight the essential need for development of highly sensitive and specific assays for gene-based diagnostic biomarkers, and to provide an overview on the most advanced gene-based potential therapeutic approaches for Parkinson disease, which are associated with the most common targets: alpha-synuclein (SNCA), leucine-rich repeat kinase-2 (LRRK2), and glucocerebrosidase (GBA).
Autophagy
Autophagy is physiologically involved in many biological processes, such as organelle turnover and intracellular homeostasis [16–18]. Pathologically, autophagic activity is altered and it is consequently involved in the pathogenesis of diseases, such as neurodegenerative diseases, including Parkinson disease [19].
Autophagy is classified into three types, which are: macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA) [11]. Macroautophagy is possibly the best-described type of autophagy, and it is referred to as autophagy throughout this review [20,21]. It starts with the formation of autophagosomes, which are double-membrane vesicles that contain the substrate to be degraded [22].
Autophagy occurs via four stages, which are induction, vesicle nucleation, autophagosome membrane elongation, and termination/fusion and degradation, and they involve autophagy-related genes (Atg) and proteins [23]. Under normal conditions, autophagy is inhibited due to the interaction of the mammalian target of rapamycin 1 (mTORC1) complex with the unc51-like kinase 1 (ULK1) complex [24]. However, this interaction stops the prevention of autophagy under stressful circumstances, such as starvation and energy depletion [25].
Activated adenosine monophosphate-activated protein kinase (AMPK) prevents mTOR resulting in the induction of autophagy [26]. The second stage in the autophagic process, which is vesicle nucleation, requires Beclin-1 complex, which consists of class III phosphoinositide 3-kinase (PI3K), p150, Beclin-1, and Atg14 [27].
Autophagosome membrane engagement is performed by the Atg12 and light chain 3 (LC3) ubiquitin-like conjugation systems [28]. Thus, ubiquitin-like Atg12 is conjugated to Atg5, Atg7, and Atg10, in which sequential reactions of E1 enzyme (Atg7) and E2 enzyme (Atg10) conjugate Atg12 to Atg5, resulting in the subsequent formation of Atg5-Atg12/Atg16L multimeric complex that localizes to the convex surface of the isolation membrane [28]. Atg5-Atg12/Atg16L complex serves as an E3 enzyme for the conjugation reaction of LC3, which is Atg8, promoting the transformation of LC3-I to LC3-II, which is essential for autophagosome formation [28]. In the last stage of autophagy, the autophagosome fuses with lysosomes, forming autophagolysosomes, so that contents of the autophagosome are degraded by hydrolases within the lysosome [29,30]. Additionally, selective forms of autophagy eliminate substrates according to the various cargoes [31]. For instance, protein aggregates are cleared by aggrephagy and damaged mitochondria are degraded by mitophagy [31,32].
In contrast, microautophagy is the direct uptake of part of the cytoplasm by lytic organelles [33,34]. Thus, the lysosomal membrane invaginates to engulf the cytoplasmic component so that it can be degraded accordingly [35]. On the other hand, CMA is a selective pathway in which the substrate to be degraded has a specific pentapeptide motif, which is identified and subsequently bound by the cytosolic heat-shock cognate 70 (HSC70) [36,37]. Consequently, it is linked to lysosome-associated membrane protein 2A (LAMP2A), leading to its translocation into the lysosomes [37].
Role of Autophagy in Parkinson Disease
ASSOCIATION BETWEEN PARKINSON DISEASE-RELATED GENE MUTATIONS AND AUTOPHAGY:
Parkinson disease is associated with genetic mutations, and a number of these genes are involved in the regulation of autophagy. Thus, recognition of the particular genes that can be mutated in association with Parkinson disease can greatly improve the understanding of the mechanisms involved in the pathogenesis of Parkinson disease. Thus, in this section, we will review the genetic mutations and their correlation with the impairment of autophagy in Parkinson disease.
ASSOCIATION BETWEEN SNCA GENE MUTATION AND AUTOPHAGY:
SNCA gene encodes α-synuclein protein, which is present chiefly in the axon terminals of presynaptic neurons [52]. Mutations and multiplications in SNCA gene encoding α-synuclein are linked to familial cases of Parkinson disease [3,53].
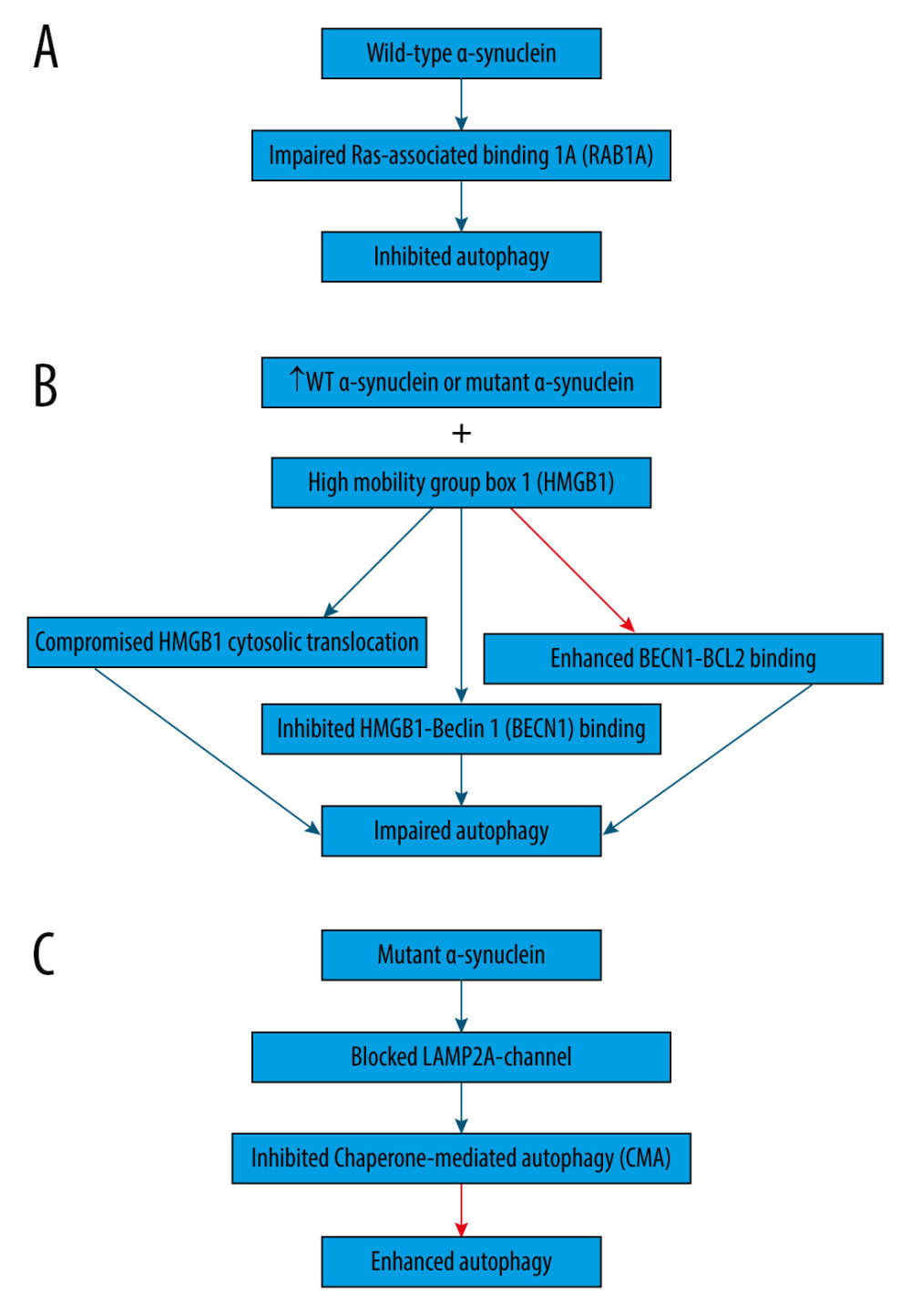
Wild-type α-synuclein is unfolded and can be sequestered via many pathways, such as autophagy and CMA [54]. On the contrary, proteins that are encoded by mutant genes can misfold and begin a vicious cycle [55]. For example, overexpression of wild-type or mutant α-synuclein can compromise the degradation pathways (Figure 1), causing abnormal buildup of α-synuclein that assembles, thus contributing to the formation of Lewy bodies in Parkinson disease brains [3,46,56]. Indeed, overexpression of wild-type α-synuclein impairs autophagy in mammalian cell lines and in transgenic mice by preventing RAB1A (Figure 1A), which is a GTPase implicated in the early secretory pathway, by causing mislocalization of the early autophagy protein ATG9 [57,58]. However, such defects may possibly be saved by RAB1A overexpression [57].
Additionally, overexpression of wild-type or mutant α-synuclein has been shown to block autophagy in PC12 cells (Figure 1B) by binding to both cytosolic and nuclear high mobility group box 1 (HMGB1) in rat PC12 cells, compromising the cytosolic translocation of HMGB1, inhibiting HMGB1-Beclin 1 (BECN1) binding, and enhancing BECN1-BCL2 binding [59]. Conversely, autophagy could be repaired by deleting HMBG1 [59].
Moreover, α-synuclein is degraded primarily by CMA due to its being identified by the HSC70 and bound to LAMP2A at the lysosomal membrane [60]. However, mutant α-synuclein inhibits CMA (Figure 1C) by acting as uptake blockers blocking LAMP2A-channel for the translocation of proteins into lysosomes [60]. Consequently, mutant α-synuclein accumulates, leading to the compensatory activation of macroautophagy in the brain of transgenic mice with overexpression of A53T-mutant α-synuclein [60].
Dopamine modifies α-synuclein making it similar to missense mutants that inhibit CMA, and thus it may further contribute to the selective susceptibility in Parkinson disease [61,62]. Furthermore, α-synuclein has been reported to interact with mitochondria and the mitophagy pathways, both directly and indirectly, since α-synuclein buildup has been correlated with the commencement of mitochondrial impairment [63]. It is possible that either α-synuclein accumulation occurs first and induces mitochondrial impairment, or mitochondrial insufficiencies cause neuronal deficits and α-synuclein build up [63]. Additionally, mutant a-synuclein has been shown to trigger mitophagy in dopaminergic neurons of a transgenic mouse model overexpressing mutant α-synuclein, where prominent mitochondrial defects and augmented autophagic cytoplasmic inclusions enclosing mitochondrial remnants were illustrated prior to the degeneration of dopaminergic neurons [64]. Moreover, overexpressing human wild-type and mutant α-synuclein in yeast cells augmented autophagy and mitophagy activities [65,66]. Under pathological circumstances, α-synuclein is translocated to the inner mitochondrial membrane [67,68]. Consequently, wild-type α-synuclein accumulates in the mitochondria of human dopaminergic neurons, suppressing mitochondrial complex I activity and promoting the formation of ROS [67,68]. Indeed, overexpression of mutant α-synuclein in dopaminergic neurons of transgenic mice led to its presence in monomeric and oligomeric forms onto the mitochondrial membranes and concomitant augmentation of mitophagy [65,69].
Moreover, incomplete autophagic breakdown of α-synuclein resulted in the formation of a truncated phosphorylated α-synuclein species called “pα-syn*”, which is hugely neurotoxic [70]. Indeed, it induces mitochondrial damage and mitophagy, and thus plays a key role in Parkinson disease pathogenesis [70]. pα-syn* has been recognized in neuronal cultures, mouse brains, and in postmortem brains from Parkinson disease patients [70].
Studies conducted in cell cultures, animal models, and human postmortem studies suggest a strong correlation between the overexpression of wild-type, mutant, or modified α-synuclein species with impaired autophagy, CMA, and mitophagy routes [46,56,70].
ASSOCIATION BETWEEN LRRK2 GENE MUTATION AND AUTOPHAGY:
LRRK2 gene encodes LRRK2 protein, which is principally present in membrane microdomains, multivesicular bodies, and autophagic vesicles [71]. Thus, LRRK2 protein is implicated in many cellular signaling pathways such as autophagy, and it possesses two different enzymatic domains, which are the kinase domain that catalyzes phosphorylation, and the Ras of complex (Roc)-GTPase domain that hydrolyses GTP-GDP [71,72]. Thus, mutations in LRRK2 can change its expression levels and/or kinase activity [73,74]. Additionally, genetic mutations in LRRK2 are associated with the majority of autosomal dominant cases of Parkinson disease [75].
Furthermore, LRRK2 has been associated with maintaining lysosomal homeostasis by its substrate Rab GTPases [76]. Thus, lysosomal overload stress causes translocation and subsequent activation of endogenous LRRK2 onto the overloaded lysosomes, where its substrates are phosphorylated and subsequently stabilized [76].
Inhibition of LRRK2 kinase reduced α-synuclein intracellular inclusions by enhancing autolysosome formation and function [77]. Autophagic dysregulation has been demonstrated in familial cases of Parkinson disease that resulted from LRRK2 mutation [78]. For instance, LRRK2-positive cytoplasmic puncta have been shown in association with autophagic vacuoles in human brain sections and in cultured human cells [79,80]. Additionally, overexpression of LC3-II has been shown in transgenic mice expressing mutant LRRK2 [81,82]. Moreover, SH-SY5Y cells expressing mutant LRRK2 displayed prominent increases in autophagic vacuoles, indicating the occurrence of autophagy, which was mediated by mitogen-activated protein kinase/extracellular signal-regulated protein kinase (MAPK/ERK) [83].
Wild-type LRRK2, in particular, can be degraded by CMA [84]. However, mutant LRRK2 or overexpressed wild-type LRRK2 can compromise CMA [84]. Indeed, mutant LRRK2 triggers LAMP2A and HSC70 buildup, obstructing its translocation at the lysosomal membrane and thus inhibiting CMA [39]. Consequently, α-synuclein breakdown was blocked, resulting in the buildup of oligomeric α-synuclein [39]. Additionally, mutant LRRK2 induced mitophagy by direct interaction with ULK1, which is required for regulating autophagy [81], and expression of mutant LRRK2 augmented mitophagy by impairing calcium homeostasis [85].
On the other hand, LRRK2 is recruited from the cytosol to the mitochondria, where wild-type LRRK2 forms a complex with the mitochondrial transport factor RHOT1/Miro1, which is an outer mitochondrial membrane protein, enhancing its elimination that precedes the initiation of mitophagy [86]. However, mutant LRRK2 disturbed this event, reducing RHOT1/Miro1 removal from damaged mitochondria and subsequently delaying mitophagy [86]. Additionally, LRRK2 mutations stop depolarization-associated mitophagy by preventing mitochondrial buildup of RAB10 [87].
ASSOCIATION BETWEEN PINK1 AND PRKN GENE MUTATION AND AUTOPHAGY:
Phosphatase and tensin homolog (PTEN)-induced kinase 1 (PINK1) and Parkin (PRKN or PARK2) proteins are functionally correlated and they control mitophagy [87]. Their homozygous or heterozygous mutations cause the most common cases of autosomal recessive early-onset Parkinson disease [88].
PINK1 kinase becomes stabilized on the outer membrane of impaired mitochondria, and it activates the E3 ubiquitin ligase PRKN to induce selective autophagy of damaged mitochondria [89]. Consequently, PRKN is triggered and recruited to the outer membrane of the impaired mitochondria, affording further Ub moieties for PINK1-dependent phosphorylation in a feed forward loop, which intensifies PINK1 and PRKN actions labeling mitochondria for mitophagy [90]. Subsequently, the mitochondria are degraded [90].
Thus, the entire loss of PINK1 function due to its mutations accompanied by heterozygous Parkinson disease-associated PRKN mutations inhibit PINK1-PRKN dependent mitophagy [91]. Consistently, reduced mitophagy or even its absence has been demonstrated in postmortem Parkinson disease brains with PINK1 or PRKN mutations [40]. Additionally, reduced mitophagy has been shown to result in the buildup of impaired mitochondria, which probably leads to neurodegeneration, particularly in dopaminergic neurons that are selectively susceptible to mitochondrial dysfunction [92].
However, Parkinson disease-like phenotypes were not shown in PINK1- or PRKN-knockdown mouse models of Parkinson disease [93]. Instead, deletion of PINK1 or PRKN in Parkinson disease mouse models caused only trivial behavioral phenotypes [93]. For example, PRKN-knockdown mice showed diminished striatal synaptic excitability and plasticity, deteriorated dopamine discharge, and nigrostriatal deficits [94]. On the other hand, PINK1 deletion significantly impaired mitochondrial respiration in the striatum and increased the striatal sensitivity to oxidative stress [95]. Consequently, mitochondrial proteins involved with energy metabolism and membrane potential were considerably changed in PINK1 knockout mice [96].
Based on its precise localization, a mutation of the PRKN protein in SH-SY5Y or HeLa cells can cause disturbance at any step of mitophagy, leading to its deterioration, which is involved in the pathogenesis of PRKN-associated parkinsonism [40].
Additionally, some Parkinson disease-linked PINK1 mutations impeded PRKN translocation, obstructing mitophagy in iPSC-derived dopaminergic neurons from Parkinson disease patients [97,98]. However, mitophagy was improved by lentiviral expression of wild-type PINK1 in the mutant iPSC-derived PINK1 neurons [99].
Stress that is induced by mitochondrial insults augmented S-nitrosylated PINK1, thus obstructing its kinase activity [98]. S-nitrosylated PINK1 impaired mitophagy by reducing PRKN translocation to mitochondrial membranes, resulting in cell death in cell lines and human iPSC-derived neurons [98]. Indeed, S-nitrosylated PINK1 has been shown in the brains of α-synuclein transgenic mice, suggesting its involvement in the pathogenesis of Parkinson disease [98].
Furthermore, PRKN function is progressively lost in dopaminergic neurons during aging and Parkinson disease [100], probably due to its being covalently changed by dopamine in substantia nigra only in the normal human brain [100], which reduces its solubility and deactivates its E3 ubiquitin ligase [100].
Altogether, the PINK1-PRKN-dependent mitophagy pathway is necessary for the elimination of impaired mitochondria, and its deficiency is possibly involved in the pathogenesis of Parkinson disease [40,88,100].
ASSOCIATION BETWEEN VPS35 GENE MUTATION AND AUTOPHAGY:
Vacuolar protein-sorting 35 (VPS35) gene encodes a subunit of the retromer complex, which mediates the retrograde transport of endosomes to the Golgi complex, thus stimulating the recycling of particular membrane proteins [101]. Additionally, retromer is required for endosomal recruitment of the Wiskott-Aldrich syndrome protein and SCAR homolog complex (WASH), which mediates protein sorting [101].
Mutations in VPS35 are associated with autosomal dominant Parkinson disease [102]. D620 mutation of VPS35 decreases its communication with the WASH complex, resulting in abnormal transferring of the autophagy protein Atg9A and impaired autophagosome formation, leading to obstruction of autophagy [103]. Furthermore, mutation of VPS35 enhances LRRK2-mediated phosphorylation of Rab protein in Parkinson disease patients [102].
Moreover, mutated or depleted VPS35 has been illustrated to impair endosome-to-Golgi retrieval of LAMP2A and increase LAMP2A breakdown, thus impairing CMA, which is essential for the degradation of α-synuclein, which subsequently builds up, leading to the development of Parkinson disease [104].
ASSOCIATION BETWEEN GBA GENE MUTATION AND AUTOPHAGY:
The GBA gene encodes glucocerebrosidase (GCase), which is a lysosomal hydrolase enzyme that cleaves glucosylceramide into glucose and ceramide [105]. Homozygous mutations in GBA gene results in Gaucher disease (GD), which is the most common sphingolipidosis lysosomal storage disorder, and it is classified into three subtypes [105]. Some patients with type 1 display parkinsonism [105].
Thus, homozygous or heterozygous mutations in GBA prevent GCase protein and lysosomal degradation, leading to α-synuclein accumulation [105,106]. Accordingly, the diminished GCase function probably disrupts the CMA pathway, augmenting α-synuclein accumulation and resulting in dopaminergic neuron degeneration [107].
ASSOCIATION BETWEEN ATP13A2 GENE MUTATION AND AUTOPHAGY:
ATP13A2 is mutated in some types of juvenile Parkinson disease [108]. It encodes a lysosomal ATPase that is suggested to be a regulator of the autophagy-lysosome pathway [109]. Consistently, ATP13A2 promotes intracellular α-synuclein accumulation by impairing lysosome exocytosis using iPSC-derived neurons from Parkinson disease patients [110]. Furthermore, ATP13A2 mutation or depletion can contribute to forms of Parkinson disease-associated neurodegeneration, since it negatively regulates another Parkinson disease-correlated gene called synaptotagmin11 (SYT11) at both transcriptional and post-translational levels, leading to mTORC1 activation and decreased levels of SYT11, which cause lysosomal dysfunction and impaired degradation of autophagosomes [109]. Additionally, ATP13A2 has been reported to facilitate the recruitment of histone deacetylase 6 (HDAC6) to lysosomes to stimulate autophagosome-lysosome fusion and subsequent autophagy [111].
Potential Therapeutic Involvement of Autophagy in Parkinson Disease
Based on the current understanding of the involvement of autophagy in association with the genetic mutations in the pathogenesis of Parkinson disease, augmenting autophagy to prevent protein aggregation and/or eliminate impaired organelles might be considered promising therapeutic approaches.
Ganoderma lucidum extract (GLE) has been shown to suppress parkinsonian phenotype by regulating both mitochondrial function and the autophagic response to oxidative stress [112]. Its administration improved the motor deficits and the selective loss of dopaminergic neurons in an MPTP model of Parkinson disease [112]. In vitro, GLE treatment reduced MPP+-induced mitochondrial dysfunction and repaired the compromised transport of impaired mitochondria, leading to enhanced autophagy in primary cultured mesencephalic neuronal cells [112]. GLE has been shown to protect neuronal cells by stimulating the activation of both the AMPK/mTOR/ULK1 and PINK1/Parkin pathways, and thus stimulating mitophagy in Parkinson disease [112]. Thus, GLE might be a promising neuroprotective agent for Parkinson disease [112].
Resveratrol has been demonstrated to induce autophagy in dopaminergic neuronal cell lines, including SH-SY5Y and PC12 cells, leading to enhanced elimination of impaired mitochondria and the degradation of mutant α-synuclein [113]. Furthermore, resveratrol has been illustrated to induce autophagic clearance of mutant α-synuclein, possibly by activating the AMPK/SIRT1 pathway, which might occur via inhibiting the mTOR pathway [113].
Rosuvastatin has been shown to ameliorate rotenone-induced neurotoxicity in SH-SY5Y cells through autophagy modulation [114]. Indeed, treatment with rosuvastatin enhanced autophagy by restoring the Beclin-1 expression, increasing the expression of AMPK, and decreasing mTOR expression in a rotenone-induced neurotoxicity model in SH-SY5Y cells [114]. Thus, rosuvastatin appears to exert neuroprotective effects on rotenone-induced dopaminergic neurotoxicity via modulating autophagy, serving as a new potential therapeutic strategy for the treatment of Parkinson disease [114].
The mammalian target of rapamycin (mTOR) is a serine/threonine protein kinase that plays a vital role in many processes, including autophagy regulation [115]. Rapamycin interferes with the assembly of mTOR, blocking its kinase activity [116]. Moreover, rapamycin improved motor dysfunction induced by 6-OHDA, probably by stimulating autophagy subsequent to the obstruction of mTORC1 activation [46]. In the wild-type SNCA transgenic mouse model, rapamycin induced autophagy and decreased the accumulation of neuronal cell bodies and synaptic regions of α-synuclein [117]. Additionally, rapamycin ameliorated the Parkinson disease phenotype of Parkin or PINK1 gene deletion in
Moxibustion has been illustrated to improve the behavioral performance and to suppress mTOR, enhancing autophagy and promoting the clearance of α-synuclein in a rat model of rotenone-induced Parkinson disease [118].
Metformin has improved motor impairment and enhanced autophagy via AMPK activation in MPTP/p Parkinson disease model mice [119].
Lithium chloride and valproic acid have been shown to enhance autophagy by suppressing inositol-monophosphatase (IMPase), which cleaves inositol-monophosphate (IMP) producing inositol (Ins), which eventually reduces autophagy [120–122].
In addition, glycogen synthase kinase3β (GSK3β) has been reported to play a chief role in methamphetamine-induced neurotoxicity by stimulating α-synuclein buildup and obstructing autophagy leading to neurodegeneration [123]. Thus, GSK3β inhibition might be considered as a possible therapeutic approach in Parkinson disease [123].
Trehalose is a natural disaccharide that inhibits protein accumulation or misfolding and promotes autophagy, thus contributing to the clearance of aggregated wild-type and mutant α-synuclein in MPTP-induced Parkinson disease [124]. Accordingly, trehalose has been shown to counteract neurotoxicity in Parkinson disease [124].
Corynoxine B is a natural autophagy inducer that repairs the deficient cytosolic translocation of HMGB1 and autophagy in cells overexpressing α-synuclein, blocking α-synuclein-HMGB1 interaction [125]. Additionally, overexpression of BECN1 or HMGB1 in cells overexpressing α-synuclein repairs autophagy and enhances the elimination of α-synuclein [125].
Geraniol is an acyclic monoterpene alcohol that is present in the essential oils of several aromatic plants [126]. Geraniol has been reported to exert a neuroprotective effect in Parkinson disease by reducing α-synuclein expression and improving autophagy [126]. Thus, geraniol has been suggested as a novel therapeutic avenue for clinical intervention in Parkinson disease [126].
Baicalein has been shown to exert neuroprotective effects in the substantia nigra of 6-hydroxydopamine-induced Parkinson disease model rats by stimulating mitochondrial autophagy via enhancing the phosphorylation level of AMPK and reducing mTOR [127].
Venlafaxine rescued dopaminergic neurons, restored the dopamine levels in the striatum, promoted autophagy, and attenuated the buildup of α-synuclein, resulting in motor recovery in rotenone-induced Parkinson disease model rats [44].
Caffeic acid (CA) has been shown to be neuroprotective, preserving dopaminergic neurons and resulting in motor recovery in a mouse model of Parkinson disease by stimulating autophagy and decreasing A53T α-synuclein [128].
However, RNA interference knockdown of
RHOT1 RNAi repairs mitophagy in neurons expressing mutant LRRK2, and protects them from oxidative stress [86]. Additionally, the neuroprotective impact of reducing RHOT1 protein levels was tested in vivo in a
Mitophagy compromised in LRRK2 mutant patient cells was repaired by deletion of the mutant LRRK2 or inhibition of LRRK2 kinase [87].
Future Developments in Gene-Based Diagnosis and Treatment of Parkinson Disease
DIAGNOSIS OF PARKINSON DISEASE:
Unfortunately, urgent medical requirements in the treatment of Parkinson disease are unmet, since no clinical biomarkers are currently available for early diagnosis of Parkinson disease, and there is no effective therapy that can interfere with the pathogenesis or progression of Parkinson disease [129].
Diagnostic biomarkers of Parkinson disease, including α-synuclein, LRRK2, and β-glucocerebrosidase (GCase), have been illustrated in biofluid studies using cerebrospinal fluid, blood cells, and urine [129]. Thus, there is a critical need to establish highly sensitive and specific assays of the specific biomarkers that are reliable for differential diagnosis [130].
Clinical assessment, neuroimaging, and electrophysiology have been helpful in the diagnosis and prognosis of Parkinson disease, since they can help in detecting the abnormalities that can precede the disease onset [131]. For instance, magnetic resonance imaging (MRI), which illustrates structural brain differences, might be useful in differentiating idiopathic Parkinson disease from other parkinsonian phenotypes [131].
Furthermore, considering the functional level, alterations in circulating oxygen levels might be useful in differentiating idiopathic Parkinson disease from a healthy state [130].
Moreover, positron emission tomography (PET) or single-photon emission computed tomography (SPECT) assesses variations among biomolecules and macromolecules, such as dopamine transporter (DAT), which is regarded as a diagnostic tool [130,132]. Additionally, electrophysiology might be helpful in assessing the neuronal activity and the subsequent monitoring of the disease [133].
However, because Parkinson disease is heterogeneous and multifactorial in its pathophysiology, the degree of the reliability of these approaches cannot be determined, and a combination of many methods and procedures may be needed [129].
POTENTIAL THERAPIES TARGETING α-SYNUCLEIN:
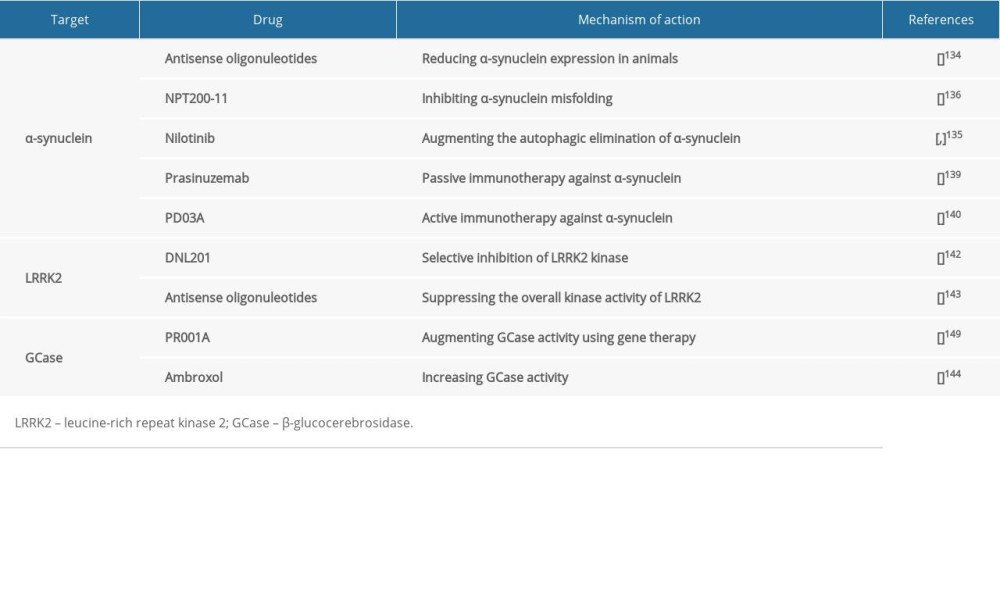
Since α-synuclein plays a key role in the pathogenesis of Parkinson disease, it is considered as a potential therapeutic target in many ongoing studies (Table 1) that aim at attenuating its burden by decreasing its gene and protein expression, repairing its proteostasis, or restricting its spread to anatomically interconnected brain regions [134].
A potential therapeutic approach that is expected to reduce α-synuclein expression is the use of antisense oligonucleotides [134], which are complimentary single-stranded RNA oligomers that can bind particular sequences within an mRNA molecule, resulting in the digestion of mRNA [134]. Preclinically, antisense oligonucleotides have been successful in reducing α-synuclein expression in animals [134].
Repairing α-synuclein proteostasis can potentially interfere with the progression of Parkinson disease, and it can be accomplished via preventing protein misfolding and accumulation or promoting its elimination [135,136]. For instance, an inhibitor of α-synuclein called NPT200-11 has been shown to be potentially useful in ameliorating α-synuclein and motor function, and completed phase I clinical trials [136]. Furthermore, nilotinib, which is an anti-cancer drug that inhibits c-Able tyrosine kinase augmenting the autophagic elimination of α-synuclein, is currently in phase 2 trials [135,137].
α-synuclein can spread among neurons, since it can be secreted extracellularly and subsequently taken up by adjacent neurons [138]. Thus, such extracellular α-synuclein has been targeted therapeutically by passive or active immunotherapy, in which α-synuclein-specific antibodies are used or synthesis of endogenous antibodies is induced consequent to the administration of modified α-synuclein [139,140]. Passive and active immunotherapies might be useful in attenuating extracellular α-synuclein and the burden of its toxic accumulation and consequently reducing its dissemination to anatomically interconnected brain areas [139,140].
Passive immunotherapy method is being attempted in clinical trials using many drugs, such as prasinuzemab, which is currently being assessed in phase II [139]. Similarly, an active immunotherapy approach has been attempted in clinical trials using drugs, including PD03A, which has completed phase 1 trials [140].
POTENTIAL THERAPIES TARGETING LRRK2:
Neuronal death caused by LRRK2 mutation in the PARK8 locus on chromosome 12 was shown to be kinase-dependent [141]. Thus, selective inhibitors of LRRK2 kinase activity were regarded as potential therapeutic candidates [142]. For instance, the selective LRRK2 kinase inhibitor, DNL201, completed a phase I clinical trial, suggesting LRRK2 kinase activity as a potential therapeutic target (Table 1) in Parkinson disease [142].
Alternatively, antisense oligonucleotides can potentially be used in suppressing the overall kinase activity of LRRK2 by obstructing LRRK2 function [143]. Indeed, LRRK2 antisense oligonucleotides have been reported to decrease Ps-129-α-synuclein inclusions by about 50% compared to controls [143]. Additionally, LRRK2 antisense oligonucleotides have been demonstrated to be useful in inhibiting Parkinson disease-associated pathology and phenotypes (Table 1) [143].
POTENTIAL THERAPIES TARGETING GCASE:
GCase is reduced in the brains of patients with Parkinson disease, particularly those with GBA1 mutations [144]. GCase insufficiency may result in the buildup of α-synuclein by maintaining oligomers, which subsequently lead to more reduction in GCase activity, resulting in a positive forward feedback loop [145,146]. Additionally, GCase inadequacy causes lysosomal impairment, leading to proteinopathy in synucleinopathies [147].
Based on their effectiveness in GD treatment, therapies like enzyme replacement therapy and glucosylceramide synthase inhibitors have been considered as potential therapeutic approaches (Table 1) in GBA-associated Parkinson disease [148]. For instance, PR001A, which augments GCase activity using gene therapy, is being assessed in a phase I safety trial [149]. Another example is ambroxol, which is an inhibitory small-molecule chaperone that increases GCase activity and decreases α-synuclein levels [144]. It is currently in phase 2 clinical trials in Parkinson disease [144].
Conclusions
There is no effective treatment that is capable of preventing neurodegeneration and disease progression in Parkinson disease. Instead of that, the current treatment for Parkinson disease is merely for symptomatic relief. The pathogenesis of Parkinson disease involves autophagy impairment. Various agents, which can stimulate the clearance of toxic α-synuclein by enhancing autophagy, could be regarded as potential therapeutic agents to halt or delay the progression of Parkinson disease. Genetic mutations associated with Parkinson disease have been shown to impact autophagy, leading to its impairment. Additionally, there is a critical need to establish highly sensitive and specific assays of the particular gene-based diagnostic biomarkers of Parkinson disease, including α-synuclein, LRRK2, and GCase, which are reliable for differential diagnosis. Furthermore, potential therapeutic targets that are gene-based in Parkinson disease are being currently developed.
References
1. Zafar S, Yaddanapudi SS: Parkinson disease, 2022, Treasure Island (FL), StatPearls
2. Liu Y, Song Y, Zhu X, MicroRNA-181a regulates apoptosis and autophagy process in Parkinson’s disease by inhibiting p38 mitogen-activated protein kinase (MAPK)/c-Jun N-terminal kinases (JNK) signaling pathways: Med Sci Monit, 2017; 23; 1597-606
3. Book A, Guella I, Candido T, A meta-analysis of alpha-synuclein multiplication in familial Parkinsonism: Front Neurol, 2018; 9; 1021
4. Cabreira V, Massano JParkinson’s disease: Clinical review and update: Acta Med Port, 2019; 32(10); 661-70 [in Portuguese]
5. Bieri G, Brahic M, Bousset L, LRRK2 modifies alpha-syn pathology and spread in mouse models and human neurons: Acta Neuropathol, 2019; 137(6); 961-80
6. Erekat NS, Apoptosis and its Role in Parkinson’s Disease: Parkinson’s disease: Pathogenesis and clinical aspects, 2018, Brisbane (AU)
7. Erekat N, Al-Jarrah MD, Interleukin-1 beta and tumor necrosis factor alpha upregulation and nuclear factor kappa B activation in skeletal muscle from a mouse model of chronic/progressive Parkinson disease: Med Sci Monit, 2018; 24; 7524-31
8. Erekat NS, Al-Jarrah MD, Association of Parkinson disease induction with cardiac upregulation of apoptotic mediators P53 and active caspase-3: An immunohistochemistry study: Med Sci Monit Basic Res, 2018; 24; 120-26
9. Lu W, Lin J, Zheng D, Overexpression of microRNA-133a inhibits apoptosis and autophagy in a cell model of Parkinson’s disease by downregulating Ras-related C3 botulinum toxin substrate 1 (RAC1): Med Sci Monit, 2020; 26; e922032
10. Erekat NS, Apoptosis and its therapeutic implications in neurodegenerative diseases: Clin Anat, 2022; 35(1); 65-78
11. Erekat NS, Programmed cell death in cerebellar Purkinje neurons: J Integr Neurosci, 2022; 21(1); 30
12. Qin Y, Qiu J, Wang P, Impaired autophagy in microglia aggravates dopaminergic neurodegeneration by regulating NLRP3 inflammasome activation in experimental models of Parkinson’s disease: Brain Behav Immun, 2021; 91; 324-38
13. Simon DK, Tanner CM, Brundin P, Parkinson disease epidemiology, pathology, genetics, and pathophysiology: Clin Geriatr Med, 2020; 36(1); 1-12
14. Ha TY, Choi YR, Noh HR, Age-related increase in caveolin-1 expression facilitates cell-to-cell transmission of alpha-synuclein in neurons: Mol Brain, 2021; 14(1); 122
15. Hardenberg MC, Sinnige T, Casford S, Observation of an alpha-synuclein liquid droplet state and its maturation into Lewy body-like assemblies: J Mol Cell Biol, 2021; 13(4); 282-94
16. Stavoe AKH, Holzbaur ELF, Autophagy in neurons: Annu Rev Cell Dev Biol, 2019; 35; 477-500
17. Levine B, Kroemer G, Biological functions of autophagy genes: A disease perspective: Cell, 2019; 176(1–2); 11-42
18. Erekat NS, Programmed cell death in diabetic nephropathy: A review of apoptosis, autophagy, and necroptosis: Med Sci Monit, 2022; 28; e937766
19. Bellomo G, Paciotti S, Gatticchi L, Parnetti L, The vicious cycle between alpha-synuclein aggregation and autophagic-lysosomal dysfunction: Mov Disord, 2020; 35(1); 34-44
20. Zhou B, Liu J, Kang R, Ferroptosis is a type of autophagy-dependent cell death: Semin Cancer Biol, 2020; 66; 89-100
21. Wang YT, Lu JH, Chaperone-mediated autophagy in neurodegenerative diseases: Molecular mechanisms and pharmacological opportunities: Cells, 2022; 11(14); 2250
22. Erekat NS, Autophagy precedes apoptosis among at risk cerebellar Purkinje cells in the shaker mutant rat: An ultrastructural study: Ultrastruct Pathol, 2018; 42(2); 162-69
23. Bomont P, GAN (gigaxonin) E3 ligase and ATG16L1: Master and commander of autophagosome production: Autophagy, 2019; 15(9); 1650-52
24. Odle RI, Walker SA, Oxley D, An mTORC1-to-CDK1 switch maintains autophagy suppression during mitosis: Mol Cell, 2020; 77(2); 228-40e7
25. Holczer M, Hajdu B, Lorincz T, Fine-tuning of AMPK-ULK1-mTORC1 regulatory triangle is crucial for autophagy oscillation: Sci Rep, 2020; 10(1); 17803
26. Zhang JA, Luan C, Huang D, Induction of autophagy by baicalin through the AMPK-mTOR pathway protects human skin fibroblasts from ultraviolet B radiation-induced apoptosis: Drug Des Devel Ther, 2020; 14; 417-28
27. Chakraborty S, Bose R, Islam S, Harnessing autophagic network is essential for trophoblast stem cell differentiation: Stem Cells Dev, 2020; 29(11); 682-94
28. Li X, He S, Ma B, Autophagy and autophagy-related proteins in cancer: Mol Cancer, 2020; 19(1); 12
29. Rost-Roszkowska MM, Vilimova J, Tajovsky K, Autophagy and apoptosis in the midgut epithelium of millipedes: Microsc Microanal, 2019; 25(4); 1004-16
30. Mendes AC, Ciccone M, Gazolla B, Bahia D, Epithelial haven and autophagy breakout in gonococci infection: Front Cell Dev Biol, 2020; 8; 439
31. Kirkin V, Rogov VV, A diversity of selective autophagy receptors determines the specificity of the autophagy pathway: Mol Cell, 2019; 76(2); 268-85
32. Deng R, Zhang HL, Huang JH, MAPK1/3 kinase-dependent ULK1 degradation attenuates mitophagy and promotes breast cancer bone metastasis: Autophagy, 2021; 17(10); 3011-29
33. Li J, Hochstrasser M, Microautophagy regulates proteasome homeostasis: Curr Genet, 2020; 66(4); 683-87
34. Schuck S, Microautophagy—distinct molecular mechanisms handle cargoes of many sizes: J Cell Sci, 2020; 133(17); jcs246332
35. Yim WW, Mizushima N, Lysosome biology in autophagy: Cell Discov, 2020; 6; 6
36. Juste YR, Cuervo AM, Analysis of Chaperone-mediated autophagy: Methods Mol Biol, 2019; 1880; 703-27
37. Kaushik S, Cuervo AM, The coming of age of chaperone-mediated autophagy: Nat Rev Mol Cell Biol, 2018; 19(6); 365-81
38. Gao F, Yang J, Wang D, Mitophagy in Parkinson’s disease: Pathogenic and therapeutic implications: Front Neurol, 2017; 8; 527
39. Ho PW, Leung CT, Liu H, Age-dependent accumulation of oligomeric SNCA/alpha-synuclein from impaired degradation in mutant LRRK2 knockin mouse model of Parkinson disease: Role for therapeutic activation of chaperone-mediated autophagy (CMA): Autophagy, 2020; 16(2); 347-70
40. Liu J, Liu W, Li R, Yang H, Mitophagy in Parkinson’s disease: From pathogenesis to treatment: Cells, 2019; 8(7); 712
41. Arotcarena ML, Teil M, Dehay B, Autophagy in synucleinopathy: The overwhelmed and defective machinery: Cells, 2019; 8(6); 565
42. Sala G, Marinig D, Arosio A, Ferrarese C, Role of Chaperone-mediated autophagy dysfunctions in the pathogenesis of Parkinson’s disease: Front Mol Neurosci, 2016; 9; 157
43. Singh SK, Dutta A, Modi G, alpha-Synuclein aggregation modulation: An emerging approach for the treatment of Parkinson’s disease: Future Med Chem, 2017; 9(10); 1039-53
44. El-Saiy KA, Sayed RH, El-Sahar AE, Kandil EA, Modulation of histone deacetylase, the ubiquitin proteasome system, and autophagy underlies the neuroprotective effects of venlafaxine in a rotenone-induced Parkinson’s disease model in rats: Chem Biol Interact, 2022; 354; 109841
45. Giorgi C, Bouhamida E, Danese A, Relevance of autophagy and mitophagy dynamics and markers in neurodegenerative diseases: Biomedicines, 2021; 9(2); 149
46. Moors TE, Hoozemans JJ, Ingrassia A, Therapeutic potential of autophagy-enhancing agents in Parkinson’s disease: Mol Neurodegener, 2017; 12(1); 11
47. Park H, Kang JH, Lee S, Autophagy in neurodegenerative diseases: A hunter for aggregates: Int J Mol Sci, 2020; 21(9); 3369
48. Jiang P, Mizushima N, Autophagy and human diseases: Cell Res, 2014; 24(1); 69-79
49. Son JH, Shim JH, Kim KH, Neuronal autophagy and neurodegenerative diseases: Exp Mol Med, 2012; 44(2); 89-98
50. Singh F, Ganley IG, Parkinson’s disease and mitophagy: An emerging role for LRRK2: Biochem Soc Trans, 2021; 49(2); 551-62
51. Hou X, Watzlawik JO, Fiesel FC, Springer W, Autophagy in Parkinson’s disease: J Mol Biol, 2020; 432(8); 2651-72
52. Liu H, Koros C, Strohaker T, A novel SNCA A30G mutation causes familial Parkinson’s disease: Mov Disord, 2021; 36(7); 1624-33
53. Day JO, Mullin S, The genetics of Parkinson’s disease and implications for clinical practice: Genes (Basel), 2021; 12(7); 1006
54. Fellner L, Gabassi E, Haybaeck J, Edenhofer F, Autophagy in α-Synucleinopathies – an overstrained system: Cells, 2021; 10(11); 3143
55. Huiting W, Bergink S, Locked in a vicious cycle: the connection between genomic instability and a loss of protein homeostasis: Genome Instability & Disease, 2021; 2(1); 1-23
56. Lei Z, Cao G, Wei G, A30P mutant alpha-synuclein impairs autophagic flux by inactivating JNK signaling to enhance ZKSCAN3 activity in midbrain dopaminergic neurons: Cell Death Dis, 2019; 10(2); 133
57. Winslow AR, Chen CW, Corrochano S, alpha-Synuclein impairs macroautophagy: Implications for Parkinson’s disease: J Cell Biol, 2010; 190(6); 1023-37
58. Lu Q, Wang PS, Yang L, Golgi-associated Rab GTPases implicated in autophagy: Cell Biosci, 2021; 11(1); 35
59. Song JX, Lu JH, Liu LF, HMGB1 is involved in autophagy inhibition caused by SNCA/alpha-synuclein overexpression: A process modulated by the natural autophagy inducer corynoxine B: Autophagy, 2014; 10(1); 144-54
60. Pupyshev AB, Korolenko TA, Akopyan AA, Suppression of autophagy in the brain of transgenic mice with overexpression of capital A, Cyrillic53capital TE, Cyrillic-mutant alpha-synuclein as an early event at synucleinopathy progression: Neurosci Lett, 2018; 672; 140-44
61. Mor DE, Tsika E, Mazzulli JR, Dopamine induces soluble alpha-synuclein oligomers and nigrostriatal degeneration: Nat Neurosci, 2017; 20(11); 1560-68
62. Martinez-Vicente M, Talloczy Z, Kaushik S, Dopamine-modified alpha-synuclein blocks chaperone-mediated autophagy: J Clin Invest, 2008; 118(2); 777-88
63. Zaltieri M, Longhena F, Pizzi M, Mitochondrial dysfunction and alpha-Synuclein synaptic pathology in Parkinson’s disease: Who’s on first?: Parkinsons Dis, 2015; 2015; 108029
64. Chen L, Xie Z, Turkson S, Zhuang X, A53T human alpha-synuclein overexpression in transgenic mice induces pervasive mitochondria macroautophagy defects preceding dopamine neuron degeneration: J Neurosci, 2015; 35(3); 890-905
65. Choubey V, Safiulina D, Vaarmann A, Mutant A53T alpha-synuclein induces neuronal death by increasing mitochondrial autophagy: J Biol Chem, 2011; 286(12); 10814-24
66. Sampaio-Marques B, Felgueiras C, Silva A, SNCA (alpha-synuclein)-induced toxicity in yeast cells is dependent on sirtuin 2 (Sir2)-mediated mitophagy: Autophagy, 2012; 8(10); 1494-509
67. Devi L, Raghavendran V, Prabhu BM, Avadhani NG, Anandatheerthavarada HK, Mitochondrial import and accumulation of alpha-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain: J Biol Chem, 2008; 283(14); 9089-100
68. Risiglione P, Zinghirino F, Di Rosa MC, Alpha-Synuclein and mitochondrial dysfunction in Parkinson’s disease: The emerging role of VDAC: Biomolecules, 2021; 11(5); 718
69. Melo TQ, van Zomeren KC, Ferrari MF, Impairment of mitochondria dynamics by human A53T alpha-synuclein and rescue by NAP (davunetide) in a cell model for Parkinson’s disease: Exp Brain Res, 2017; 235(3); 731-42
70. Grassi D, Howard S, Zhou M, Identification of a highly neurotoxic alpha-synuclein species inducing mitochondrial damage and mitophagy in Parkinson’s disease: Proc Natl Acad Sci USA, 2018; 115(11); E2634-43
71. Alegre-Abarrategui J, Christian H, Lufino MM, LRRK2 regulates autophagic activity and localizes to specific membrane microdomains in a novel human genomic reporter cellular model: Hum Mol Genet, 2009; 18(21); 4022-34
72. Liu Z, Mobley JA, DeLucas LJ, Kahn RA, West AB, LRRK2 autophosphorylation enhances its GTPase activity: FASEB J, 2016; 30(1); 336-47
73. Henry AG, Aghamohammadzadeh S, Samaroo H, Pathogenic LRRK2 mutations, through increased kinase activity, produce enlarged lysosomes with reduced degradative capacity and increase ATP13A2 expression: Hum Mol Genet, 2015; 24(21); 6013-28
74. Reinhardt P, Schmid B, Burbulla LF, Genetic correction of a LRRK2 mutation in human iPSCs links parkinsonian neurodegeneration to ERK-dependent changes in gene expression: Cell Stem Cell, 2013; 12(3); 354-67
75. Marchand A, Drouyer M, Sarchione A, LRRK2 phosphorylation, more than an epiphenomenon: Front Neurosci, 2020; 14; 527
76. Ysselstein D, Nguyen M, Young TJ, LRRK2 kinase activity regulates lysosomal glucocerebrosidase in neurons derived from Parkinson’s disease patients: Nat Commun, 2019; 10(1); 5570
77. Obergasteiger J, Frapporti G, Lamonaca G, Kinase inhibition of G2019S-LRRK2 enhances autolysosome formation and function to reduce endogenous alpha-synuclein intracellular inclusions: Cell Death Discov, 2020; 6; 45
78. Madureira M, Connor-Robson N, Wade-Martins R, LRRK2: Autophagy and lysosomal activity: Front Neurosci, 2020; 14; 498
79. Beilina A, Cookson MR, Genes associated with Parkinson’s disease: Regulation of autophagy and beyond: J Neurochem, 2016; 139(Suppl 1); 91-107
80. Albanese F, Novello S, Morari M, Autophagy and LRRK2 in the aging brain: Front Neurosci, 2019; 13; 1352
81. Zhu Y, Wang C, Yu M, ULK1 and JNK are involved in mitophagy incurred by LRRK2 G2019S expression: Protein Cell, 2013; 4(9); 711-21
82. Toyofuku T, Morimoto K, Sasawatari S, Kumanogoh A, Leucine-rich repeat kinase 1 regulates autophagy through turning on TBC1D2-dependent Rab7 inactivation: Mol Cell Biol, 2015; 35(17); 3044-58
83. Plowey ED, Cherra SJ, Liu YJ, Chu CT, Role of autophagy in G2019S-LRRK2-associated neurite shortening in differentiated SH-SY5Y cells: J Neurochem, 2008; 105(3); 1048-56
84. Orenstein SJ, Kuo SH, Tasset I, Interplay of LRRK2 with chaperone-mediated autophagy: Nat Neurosci, 2013; 16(4); 394-406
85. Cherra SJ, Steer E, Gusdon AM, Mutant LRRK2 elicits calcium imbalance and depletion of dendritic mitochondria in neurons: Am J Pathol, 2013; 182(2); 474-84
86. Wang X, Destructive cellular paths underlying familial and sporadic Parkinson disease converge on mitophagy: Autophagy, 2017; 13(11); 1998-99
87. Wauters F, Cornelissen T, Imberechts D, LRRK2 mutations impair depolarization-induced mitophagy through inhibition of mitochondrial accumulation of RAB10: Autophagy, 2020; 16(2); 203-22
88. Vizziello M, Borellini L, Franco G, Ardolino G, Disruption of mitochondrial homeostasis: The role of PINK1 in Parkinson’s disease: Cells, 2021; 10(11); 3022
89. Kane LA, Lazarou M, Fogel AI, PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity: J Cell Biol, 2014; 205(2); 143-53
90. Koyano F, Yamano K, Kosako H, Parkin recruitment to impaired mitochondria for nonselective ubiquitylation is facilitated by MITOL: J Biol Chem, 2019; 294(26); 10300-14
91. Clark EH, Vázquez de la Torre A, Hoshikawa T, Briston T, Targeting mitophagy in Parkinson’s disease: J Biol Chem, 2021; 296; 100209
92. Cen X, Zhang M, Zhou M, Mitophagy regulates neurodegenerative diseases: Cells, 2021; 10(8); 1876
93. Chia SJ, Tan EK, Chao YX, Historical perspective: Models of Parkinson’s disease: Int J Mol Sci, 2020; 21(7); 2464
94. Kitada T, Pisani A, Porter DR, Impaired dopamine release and synaptic plasticity in the striatum of PINK1-deficient mice: Proc Natl Acad Sci USA, 2007; 104(27); 11441-46
95. Stauch KL, Villeneuve LM, Purnell PR, Loss of Pink1 modulates synaptic mitochondrial bioenergetics in the rat striatum prior to motor symptoms: Concomitant complex I respiratory defects and increased complex II-mediated respiration: Proteomics Clin Appl, 2016; 10(12); 1205-17
96. Bus C, Zizmare L, Feldkaemper M, Human dopaminergic neurons lacking PINK1 exhibit disrupted dopamine metabolism related to vitamin B6 co-factors: iScience, 2020; 23(12); 101797
97. Chung SY, Kishinevsky S, Mazzulli JR, Parkin and PINK1 patient iPSC-derived midbrain dopamine neurons exhibit mitochondrial dysfunction and alpha-Synuclein accumulation: Stem Cell Reports, 2016; 7(4); 664-77
98. Oh CK, Sultan A, Platzer J, S-nitrosylation of PINK1 attenuates PINK1/Parkin-dependent mitophagy in hiPSC-based Parkinson’s disease models: Cell Rep, 2017; 21(8); 2171-82
99. Seibler P, Graziotto J, Jeong H, Mitochondrial Parkin recruitment is impaired in neurons derived from mutant PINK1 induced pluripotent stem cells: J Neurosci, 2011; 31(16); 5970-76
100. Tokarew JM, El-Kodsi DN, Lengacher NA, Age-associated insolubility of parkin in human midbrain is linked to redox balance and sequestration of reactive dopamine metabolites: Acta Neuropathol, 2021; 141(5); 725-54
101. Williams ET, Chen X, Moore DJ, VPS35, the retromer complex and Parkinson’s disease: J Parkinsons Dis, 2017; 7(2); 219-33
102. Mir R, Tonelli F, Lis P, The Parkinson’s disease VPS35[D620N] mutation enhances LRRK2-mediated Rab protein phosphorylation in mouse and human: Biochem J, 2018; 475(11); 1861-83
103. Zavodszky E, Seaman MN, Moreau K, Mutation in VPS35 associated with Parkinson’s disease impairs WASH complex association and inhibits autophagy: Nat Commun, 2014; 5; 3828
104. Tang FL, Erion JR, Tian Y, VPS35 in dopamine neurons is required for endosome-to-golgi retrieval of Lamp2a, a receptor of chaperone-mediated autophagy that is critical for alpha-Synuclein degradation and prevention of pathogenesis of Parkinson’s disease: J Neurosci, 2015; 35(29); 10613-28
105. Smith L, Schapira AHV, GBA variants and Parkinson disease: Mechanisms and treatments: Cells, 2022; 11(8); 1261
106. Du TT, Wang L, Duan CL, GBA deficiency promotes SNCA/alpha-synuclein accumulation through autophagic inhibition by inactivated PPP2A: Autophagy, 2015; 11(10); 1803-20
107. Kuo SH, Tasset I, Cheng MM, Mutant glucocerebrosidase impairs alpha-synuclein degradation by blockade of chaperone-mediated autophagy: Sci Adv, 2022; 8(6); eabm6393
108. Suleiman J, Hamwi N, El-Hattab AW, ATP13A2 novel mutations causing a rare form of juvenile-onset Parkinson disease: Brain Dev, 2018; 40(9); 824-26
109. Bento CF, Ashkenazi A, Jimenez-Sanchez M, Rubinsztein DC, The Parkinson’s disease-associated genes ATP13A2 and SYT11 regulate autophagy via a common pathway: Nat Commun, 2016; 7; 11803
110. Sepulveda D, Cisternas-Olmedo M, Arcos J, Contribution of autophagy-lysosomal pathway in the exosomal secretion of alpha-Synuclein and its impact in the progression of Parkinson’s disease: Front Mol Neurosci, 2022; 15; 805087
111. Wang R, Tan J, Chen T, ATP13A2 facilitates HDAC6 recruitment to lysosome to promote autophagosome-lysosome fusion: J Cell Biol, 2019; 218(1); 267-84
112. Ren ZL, Wang CD, Wang T, Ganoderma lucidum extract ameliorates MPTP-induced parkinsonism and protects dopaminergic neurons from oxidative stress via regulating mitochondrial function, autophagy, and apoptosis: Acta Pharmacol Sin, 2019; 40(4); 441-50
113. Wu Y, Li X, Zhu JX, Resveratrol-activated AMPK/SIRT1/autophagy in cellular models of Parkinson’s disease: Neurosignals, 2011; 19(3); 163-74
114. Kang SY, Lee SB, Kim HJ, Autophagic modulation by rosuvastatin prevents rotenone-induced neurotoxicity in an in vitro model of Parkinson’s disease: Neurosci Lett, 2017; 642; 20-26
115. Yoon MS, The role of mammalian target of rapamycin (mTOR) in insulin signaling: Nutrients, 2017; 9(11); 1176
116. Waldner M, Fantus D, Solari M, Thomson AW, New perspectives on mTOR inhibitors (rapamycin, rapalogs and TORKinibs) in transplantation: Br J Clin Pharmacol, 2016; 82(5); 1158-70
117. Blanz J, Saftig P, Parkinson’s disease: Acid-glucocerebrosidase activity and alpha-synuclein clearance: J Neurochem, 2016; 139(Suppl 1); 198-215
118. Wang SJ, Wang Q, Ma J, Effect of moxibustion on mTOR-mediated autophagy in rotenone-induced Parkinson’s disease model rats: Neural Regen Res, 2018; 13(1); 112-18
119. Lu M, Su C, Qiao C, Metformin prevents dopaminergic neuron death in MPTP/P-induced mouse model of Parkinson’s disease via autophagy and mitochondrial ROS clearance: Int J Neuropsychopharmacol, 2016; 19(9); pyw047
120. Hou L, Xiong N, Liu L, Lithium protects dopaminergic cells from rotenone toxicity via autophagy enhancement: BMC Neurosci, 2015; 16; 82
121. Sarkar S, Floto RA, Berger Z, Lithium induces autophagy by inhibiting inositol monophosphatase: J Cell Biol, 2005; 170(7); 1101-11
122. Li XZ, Chen XP, Zhao K, Therapeutic effects of valproate combined with lithium carbonate on MPTP-induced parkinsonism in mice: Possible mediation through enhanced autophagy: Int J Neurosci, 2013; 123(2); 73-79
123. Li L, Chen S, Wang Y, Role of GSK3beta/alpha-synuclein axis in methamphetamine-induced neurotoxicity in PC12 cells: Toxicol Res (Camb), 2018; 7(2); 221-34
124. Khalifeh M, Barreto GE, Sahebkar A, Trehalose as a promising therapeutic candidate for the treatment of Parkinson’s disease: Br J Pharmacol, 2019; 176(9); 1173-89
125. Song JX, Lu JH, Liu LF, B1 is involved in autophagy inhibition caused by SNCA/a-synuclein overexpression: A process modulated by the natural autophagy inducer corynoxine B: Autophagy, 2015; 11(9); 1708
126. Rekha KR, Inmozhi Sivakamasundari R, Geraniol protects against the protein and oxidative stress induced by rotenone in an in vitro model of Parkinson’s disease: Neurochem Res, 2018; 43(10); 1947-62
127. Chen M, Peng L, Gong P, Baicalein induces mitochondrial autophagy to prevent Parkinson’s disease in rats via miR-30b and the SIRT1/AMPK/mTOR pathway: Front Neurol, 2021; 12; 646817
128. Zhang Y, Wu Q, Zhang L, Caffeic acid reduces A53T alpha-synuclein by activating JNK/Bcl-2-mediated autophagy in vitro and improves behaviour and protects dopaminergic neurons in a mouse model of Parkinson’s disease: Pharmacol Res, 2019; 150; 104538
129. Polissidis A, Petropoulou-Vathi L, Nakos-Bimpos M, Rideout HJ, The future of targeted gene-based treatment strategies and biomarkers in Parkinson’s disease: Biomolecules, 2020; 10(6); 912
130. He R, Yan X, Guo J, Recent advances in biomarkers for Parkinson’s disease: Front Aging Neurosci, 2018; 10; 305
131. Pagano G, Niccolini F, Politis M, Imaging in Parkinson’s disease: Clin Med (Lond), 2016; 16(4); 371-75
132. Akdemir UO, Tokcaer AB, Karakus A, Kapucu LO, Brain 18F-FDG PET imaging in the differential diagnosis of parkinsonism: Clin Nucl Med, 2014; 39(3); e220-26
133. Jackson N, Cole SR, Voytek B, Swann NC, Characteristics of waveform shape in Parkinson’s disease detected with scalp electroencephalography: eNeuro, 2019; 6(3) ENEURO.0151-19.2019
134. Cole TA, Zhao H, Collier TJ, alpha-Synuclein antisense oligonucleotides as a disease-modifying therapy for Parkinson’s disease: JCI Insight, 2021; 6(5); e135633
135. Hebron ML, Lonskaya I, Moussa CE, Nilotinib reverses loss of dopamine neurons and improves motor behavior via autophagic degradation of alpha-synuclein in Parkinson’s disease models: Hum Mol Genet, 2013; 22(16); 3315-28
136. Price DL, Koike MA, Khan A, The small molecule alpha-synuclein misfolding inhibitor, NPT200-11, produces multiple benefits in an animal model of Parkinson’s disease: Sci Rep, 2018; 8(1); 16165
137. Pagan F, Hebron M, Valadez EH, Nilotinib effects in Parkinson’s disease and dementia with Lewy bodies: J Parkinsons Dis, 2016; 6(3); 503-17
138. Brundin P, Dave KD, Kordower JH, Therapeutic approaches to target alpha-Synuclein pathology: Exp Neurol, 2017; 298(Pt B); 225-35
139. Jankovic J, Goodman I, Safirstein B, Safety and tolerability of multiple ascending doses of PRX002/RG7935, an anti-alpha-Synuclein monoclonal antibody, in patients with Parkinson disease: A randomized clinical trial: JAMA Neurol, 2018; 75(10); 1206-14
140. Poewe W, Volc D, Seppi K, Safety and tolerability of active immunotherapy targeting alpha-Synuclein with PD03A in patients with early Parkinson’s disease: A randomized, placebo-controlled, phase 1 study: J Parkinsons Dis, 2021; 11(3); 1079-89
141. Smith WW, Pei Z, Jiang H, Kinase activity of mutant LRRK2 mediates neuronal toxicity: Nat Neurosci, 2006; 9(10); 1231-33
142. Jennings D, Huntwork-Rodriguez S, Henry AG, Preclinical and clinical evaluation of the LRRK2 inhibitor DNL201 for Parkinson’s disease: Sci Transl Med, 2022; 14(648); eabj2658
143. Zhao HT, John N, Delic V, LRRK2 antisense oligonucleotides ameliorate alpha-Synuclein inclusion formation in a Parkinson’s disease mouse model: Mol Ther Nucleic Acids, 2017; 8; 508-19
144. Mullin S, Smith L, Lee K, Ambroxol for the treatment of patients with Parkinson disease with and without glucocerebrosidase gene mutations: A nonrandomized, noncontrolled trial: JAMA Neurol, 2020; 77(4); 427-34
145. Mazzulli JR, Xu YH, Sun Y, Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies: Cell, 2011; 146(1); 37-52
146. Lwin A, Orvisky E, Goker-Alpan O, Glucocerebrosidase mutations in subjects with parkinsonism: Mol Genet Metab, 2004; 81(1); 70-73
147. Sardi SP, Cheng SH, Shihabuddin LS, Gaucher-related synucleinopathies: The examination of sporadic neurodegeneration from a rare (disease) angle: Prog Neurobiol, 2015; 125; 47-62
148. Sardi SP, Clarke J, Viel C, Augmenting CNS glucocerebrosidase activity as a therapeutic strategy for parkinsonism and other Gaucher-related synucleinopathies: Proc Natl Acad Sci USA, 2013; 110(9); 3537-42
149. Abeliovich A, Hefti F, Sevigny J, Gene therapy for Parkinson’s disease associated with GBA1 mutations: J Parkinsons Dis, 2021; 11(s2); S183-88
In Press
Meta-Analysis
Effectiveness of Cardiac Telerehabilitation in Improving Functional Capacity, Quality Of Life and Cardiovas...Med Sci Monit In Press; DOI: 10.12659/MSM.953366
Clinical Research
Analysis of the Clinical Characteristics and Endoscopic Features of Phytobezoar-Induced Ulcers and Gastric ...Med Sci Monit In Press; DOI: 10.12659/MSM.952191
Clinical Research
Effect of Indirect Co-Culture With Gingival Mesenchymal Stem Cells on Cytokine Secretion in Primary Oral Sq...Med Sci Monit In Press; DOI: 10.12659/MSM.952439
Clinical Research
Comparison of Sleep Architecture in Individuals Aged 65 to 80 Years With and Without Mild Cognitive Impairm...Med Sci Monit In Press; DOI: 10.12659/MSM.952493
Most Viewed Current Articles
17 Jan 2024 : Review article 14,176,468
Vaccination Guidelines for Pregnant Women: Addressing COVID-19 and the Omicron VariantDOI :10.12659/MSM.942799
Med Sci Monit 2024; 30:e942799
13 Nov 2021 : Clinical Research 3,759,966
Acceptance of COVID-19 Vaccination and Its Associated Factors Among Cancer Patients Attending the Oncology ...DOI :10.12659/MSM.932788
Med Sci Monit 2021; 27:e932788
14 Dec 2022 : Clinical Research 2,466,248
Prevalence and Variability of Allergen-Specific Immunoglobulin E in Patients with Elevated Tryptase LevelsDOI :10.12659/MSM.937990
Med Sci Monit 2022; 28:e937990
16 May 2023 : Clinical Research 708,898
Electrophysiological Testing for an Auditory Processing Disorder and Reading Performance in 54 School Stude...DOI :10.12659/MSM.940387
Med Sci Monit 2023; 29:e940387