18 August 2021: Database Analysis
Genome-Wide Profiling of Alternative Splicing Signatures Associated with Prognosis and Immune Microenvironment of Hepatocellular Carcinoma
Zhikun Liu12ABCDEF, Jiangwei Ye13ABCDE, Abid Ali Khan12ABCDEF, Jun Chen12BCDE, Lin Zhou2BCDE, Shusen Zheng2ADE, Xiao Xu12ADG*DOI: 10.12659/MSM.930052
Med Sci Monit 2021; 27:e930052






Abstract
BACKGROUND: The potential roles of alternative splicing (AS) in HCC remain unknown. This study aimed to identify AS signatures associated with the prognosis that influence the immune microenvironment of HCC.
MATERIAL AND METHODS: The SpliceSeq tool was employed for genome-wide profiling of 7 AS events in 361 HCC patients from The Cancer Genome Atlas (TCGA). A prognostic signature was built by integrating Cox regression and the least absolute shrinkage and selection operator (LASSO). The support vector machine (SVM) and receiver operating characteristic curve (ROC) were employed to analyze the AS events in the signatures to discriminate the immune microenvironment.
RESULTS: There were 3546 AS events highly linked to the survival of patients with HCC. The AS signature could effectively stratify HCC patients. Clustering analysis revealed 3 different immune clusters characterized with significantly different prognoses and were significantly correlated with AS signatures. The AS events in the final prognostic signature classified the immune cluster with an average AUC of the ROC (0.88). Moreover, a potential regulatory network of splicing events in HCC is presented.
CONCLUSIONS: We established the prognostic signature based on AS, which can effectively stratify HCC patients and predict the immune subtypes. Moreover, novel RNA splicing patterns and splicing-regulatory networks involved in HCC were discovered.
Keywords: Alternative Splicing, Carcinoma, Hepatocellular, tumor microenvironment, Biomarkers, Tumor, Cluster Analysis, Databases, Genetic, Gene Regulatory Networks, Genome-Wide Association Study, Liver Neoplasms, Proportional Hazards Models, RNA Splice Sites, RNA Splicing Factors, ROC Curve, support vector machine
Background
Protein-coding genes are finite. Hence, they cannot create huge protein diversities that are essential for the complex functional and regulatory processes in eukaryotic cells. Alternative splicing (AS) of pre-mRNA is one of the pathways that lead to protein diversity. It does so by creating mRNA isoforms from the same set of genes. In the process, exons and introns are removed from the human multi-exon/introns genes [1,2]. AS also down-regulates the translation of mRNA isoforms by introducing a premature stop codon [3]. Under normal physiological circumstances, alternative mRNA splicing is a well-regulated process, orchestrated by a small number of splicing factors (SFs). However, abrupt disruption in the AS process or mutation in genes encoding for SFs may occur and lead to cancer [4]. High-throughput sequencing technologies have brought immense excitement to cancer genome research, and the link between splicing defects in oncogenic protein isoforms has been the focus of extensive research during the last decade [5,6]. Indeed, AS events influence immune escape, proliferation, hypoxia, apoptosis, angiogenesis, and metastasis of tumor cells. Furthermore, diagnostic and prognostic values of AS in cancer are becoming more evident form recent research [7].
Liver cancers are highly prevalent globally. The most common among them is hepatocellular carcinoma (HCC), which constitutes 90% of all primary liver cancers [8]. HCC recurrence after surgery is a commonly encountered problem in clinical practice [9,10]. Prognostic biomarkers, overall survival, and recurrence-free survival are some of the indicators of its aggressiveness. However, no clear-cut molecular predictive biomarker is available. With recent progress in the use of high-throughput sequencing technologies, bioinformatics, and online availability of wide cancer genome data, systematic genome-wide analysis of different cancers has become possible [11]. Several studies have been carried out recently to identify cancer-associated AS events using The Cancer Genome Atlas (TCGA). Moreover, these data have been further utilized to construct prognostic signatures [12,13].
Several studies have been conducted to study AS events in HCC, suggesting that alteration of AS is a significant process involved in oncogenesis and could be used as a key prognostic biomarker in HCC [15–18]. In the present study, an AS sequence database of 361 HCC patients was extracted from TCGA, and several prognosis-associated AS events were systematically uncovered. The AS events associated with the immune microenvironment were investigated first in HCC. We used machine learning methods to construct an AS-based model for prognosis (LASSO Cox) and to discriminate the immune microenvironment (SVM). The
Material and Methods
DATA ACQUISITION FROM TCGA:
TCGA SpliceSeq is an online data resource that provides AS profiles of 33 different tumors [19]. mRNA splicing patterns of TCGA samples were analyzed using the SpliceSeq application [20]. RNAseq and clinical data of 361 HCC patients were downloaded from TCGA data domain (https://portal.gdc.cancer.gov/). SpliceSeq tool, a java program application, was used to analyze the mRNA splicing patterns of TCGA samples [21]. Seven AS types with a percent spliced in (PSI) value of more than 75% (value range from 0% to 100%) in HCC were retrieved from the TCGA SpliceSeq database. The PSI value indicates the efficiency of splicing a specific exon into the transcript population of a gene. It is the ratio between reads including or excluding exons [20]. The 7 AS events were: alternate acceptor site (AA), alternate donor site (AD), alternate terminator (AT), alternate promoter (AP), retained intron (RI), mutually exclusive exon (ME), and exon skip (ES).
SURVIVAL-ASSOCIATED AS EVENTS IN HCC:
The correlation of AS events to RFS and OS was explored using univariate Cox regression analysis. The UpSet plot visualization technique was utilized to quantitatively analyze interactive sets and intersections between the 7 types of AS events. Prognostic signatures were created with the LASSO tool, which was also used for Cox regression survival analysis, given that it is fit for high-dimensional data [22]. The top 50 key AS events in RI, ME, ES, AA, AT, AD, and AP that were linked to survival were selected to construct the prognostic signatures by LASSO Cox based on the minimum criteria. They were then combined to construct the final prognostic signature using the same method. “Glmnet” package in R was utilized to estimate the coefficients and partial likelihood deviance.
BIOINFORMATICS:
The immune and non-immune cell populations were quantified from bulk gene expression data of HCC tumors. Immune cell types in the tumor microenvironment were identified using single-sample gene set enrichment analysis (ssGSEA) as described by Charoentong [23]. Specific cell-type transcriptomic signatures were used as reported previously [23–25]. The average score of the cell was used in cases where there were common cell types among different methods. Cluster testing was performed to determine the optimal number of stable HCC microenvironment subtypes, then silhouette analysis was conducted to validate the stability of the clustering. Unsupervised cluster analysis was then performed to cluster samples based on the constituent pattern of all the cell types. The k-means clustering result was utilized to reorder the samples and scale the original cell scores before heatmap plotting. The AS events in the final signatures were used to construct the model to discriminate the different immune clusters by SVM. The risk scores were calculated from the final prognostic signatures and separated into the high and low groups using the median. The clinical parameters of patients were then integrated into the multivariable Cox regression analysis to create a final signature with the potential to serve as an independent prognostic predictor.
SF-AS REGULATORY NETWORK:
Expression data of SF genes in HCC were derived from the TCGA database. Spearman tests were carried out to explore the association between the PSI values of survival-associated AS and expression of SF genes. The “Benjamini & Hochberg” method was employed to adjust
STATISTICAL ANALYSIS:
The R version 3.6.2. software was employed for data analysis. Kaplan-Meier curves of survival probability were compared with the log-rank test using “survminer”. The LASSO Cox regression model was employed to assess ideal coefficient for each variable and to estimate the deviance likelihood via 1-standard error criteria using “glmnet”. All tests were two-sided, and
Results
INTEGRATED AS EVENTS PROFILES IN HCC PATIENTS:
Integrated mRNA splicing events profiles of 361 HCC patients were explored using the data obtained from TCGA. We detected 34 163 mRNA splicing events in 7196 genes. This indicated that the average number of AS events per gene was 4 to 5. Of note, ES was the most frequent splice signature, followed by AP and AT, respectively. We found 6965 ESs in 2954 genes, 2263 RIs in 1561 genes, 5346 APs in 2167 genes, 4892 ATs in 2148 genes, 2331 ADs in 1663 genes, 2666 AAs in 1937 genes, and 137 MEs in 135 genes. The genes and AS events of each AS type are shown in Figure 1.
The survival-associated AS events in HCC patients were then investigated. One survival-associated gene had 2 or more AS events. The UpSet plots further revealed that 1 gene had up to 4 types of AS events, which were correlated with overall survival and recurrence-free survival (Figure 2), with ES being the most frequent survival-associated AS event.
AS PROGNOSTIC SIGNATURES FOR HCC PATIENTS:
LASSO Cox analyses of overall survival were used to assess the prognostic significance of AS events in HCC patients following the univariate Cox analysis. The top 50 most significant survival-associated AS events in AA, AD, AP, AT, ES, and RI were used to develop prognostic signatures by LASSO Cox based on the minimum criteria (Figures 3, 4). Finally, the top 50 most significant survival-associated AS events in the 7 types were used to construct the final prognostic signature. LASSO Cox analysis failed for ME because of lack of enough ME events in HCC patients. A risk score was then obtained using coefficients from the LASSO algorithm. The HCC patients were classified into high- and low-risk groups on the basis of their median risk scores to further investigate the prognostic role of the risk signatures. Significant differences in overall survival (Figure 5) and in recurrence-free survival (Supplementary Figure 1) were observed. The final prognostic signature had excellent ability to distinguish HCC patients with different clinical outcomes (Figure 5G). The sets of genes and splice events for each type of AS are submitted as the supplementary data file (Supplementary Table 1).
AS ASSOCIATED WITH THE IMMUNE MICROENVIRONMENT:
To query the relationship between AS events and the tumor immune microenvironment, several immune and non-immune cell populations were quantified from the bulk gene expression data of HCC patients. A total of 43 cell populations grouped into adaptive immune cells, innate immune cells, and stromal cells were identified. The immune cell types and the proportional value are shown in Supplementary Table 2. Unsupervised cluster analysis using all the 43 cell populations showed 3 different patterns of microenvironment cell types in HCC patients (Figure 6A). Three was the optimal and stable clustering number (Supplementary Figure 2). Cluster 1 (immune-high subtype, “hot tumor”) was characterized by increased B cells, T cells, and other immune and non-immune cell types. Cluster 2 (Immune-mid subtype) showed moderately increased innate immune cell infiltration, with lesser adaptive immune cell infiltration. Cluster 3 (Immune-low subtype, “immune-desert tumor”) showed low immune and non-immune cell infiltration. The association between different type prognostic signatures and immune clusters was then explored. All the AS signatures were found to be significantly correlated with the immune clusters. In addition, the immune-high subtype was found to capture 2 distinct subtypes: active (cluster 1A) and exhausted (cluster 1B) subtypes. The exhausted subtype had increased Treg and Th2 cells, which confirmed their diverse roles in immune exhaustion. Moreover, there were significant differences in RFS (Figure 6B) and OS (Supplementary Figure 3) between the different immune-cluster subtypes. The ability of the AS events to predict the immune clusters in the final prognostic signatures was further confirmed using the ROC curves (Figure 7). The AUC of the average ROC reached 0.88.
Univariate Cox analysis was then performed to assess the effect of clinical characteristics, the risk score, and immune clusters on RFS and OS. The univariate analyses of RFS and OS are shown in Supplementary Tables 3 and 4, respectively. Multivariate Cox analysis was then further performed for RFS (Figure 8A) and OS (Figure 8B). The risk score calculated from the final prognostic signature, the immune clusters, and stage were found to be independent prognostic indicators.
SURVIVAL-ASSOCIATED SF-AS NETWORK:
SFs regulate RNA splicing by binding to the cis-regulatory elements in pre-mRNA [26]. A splicing-regulatory network of key AS events related to the survival in HCC patients was therefore constructed to determine whether SFs with altered expression were potential regulators of significant AS events. We identified 49 SFs as having a significant association with OS in HCC patients based on their expression levels. Spearman correlation analyses comparing gene expression of survival-associated SFs and PSI values of survival-associated AS events were further performed. Only significant correlations (P<0.001) are presented in Figure 9A. In the correlation network, 26 survival-associated SFs (purple dots) were significantly related to 91 survival-associated AS events. Among these 91, 48 (green dots) were notably linked to favorable survival while 43 (red dots) were markedly correlated with poor survival in HCC patients (Figure 9A). The network further revealed a positive correlation (represented with red lines) between the red and purple dots (ie, the poorest survival prognostic AS events). Of note, a negative relationship was observed between the best survival-associated AS events and the expression of SFs. Specifically, splicing factor SRSF2 was markedly correlated with poor overall survival and disease-free survival (Figure 9B). Correlations between AP PSI values of CAPRIN1 and ES PSI values of SULT1A2 and splicing factor SRSF2 are shown in dot plots (Figure 9C).
Discussion
AS is an essential mechanism for creating huge protein diversity in eukaryotic cells. Although it is a well-regulated mechanism, disturbances in the mRNA AS process can occur and promote tumorigenesis [27]. Dysregulation in AS can lead to the formation of cancer-promoting protein isoforms associated with proliferation, metastasis, and drug resistance. For example, CD44 isoforms are critical in colorectal cancer initiation [28]. Several aspects of AS abnormalities such as the production of cancer-specific isoform of certain genes, inactivation of tumor suppressor genes, activation of oncogenes, disturbance of cell regulatory pathways, and mutation of SFs promote tumorigenesis. Recent genome-wide data analysis using bioinformatics tools have revealed hundreds of aberrant AS events strongly related to the prognosis of NSCLC and thyroid cancer [29,30].
Liver cancers are highly prevalent globally. The most common among them is HCC, which constitutes 90% of all primary liver cancers [8]. Predictive molecular biomarkers such as AFP, micro RNAs, Wnt5a, and human carbonyl reductase 2, among others, are supplemented by imaging techniques to diagnose and predict the likelihood of HCC occurrence [31]. However, the predictive capability of these biomarkers is limited [32]. Owing to its global burden, the development of new methods to identify more diagnostic and predictive biomarkers for HCC is thus crucial. Recent advances in high-throughput sequencing technologies and bioinformatics tools have enabled comprehensive analysis and characterization of genetic aberrations of the spliceosome and splice sites to be possible [33]. The prognostic and diagnostic values of AS signatures have been previously revealed in different studies. Variation in AS signatures and cancer-specific AS events could be used as prognostic, predictive, and diagnostic biomarkers for different types of cancers. SpliceSeq analyses have been previously done to generate alterative splicing profiles for different cancers and to identify significant splicing events that can predict cancer prognosis [26,34]. Genome-wide analysis to generate AS profiles of HCC patients followed by identification of significant AS events to predict prognosis has also been done in several studies [15–18]. Compared with these previous, the major original features of the present study are: we associated the AS events with the immune microenvironment of HCC, and we used machine learning methods to construct an AS-based model for prognosis (LASSO Cox) and to discriminate the immune microenvironment (SVM). The final prognostic signatures had excellent ability to distinguish HCC patients with different clinical outcomes. The relationship between AS events and the tumor immune microenvironment was explored, and significant correlations between AS signature and immune clusters were identified. An AS to SF correlation network was constructed to give further insights into the regulatory role of SF in AS events in HCC patients. High expression of SRSF2 was found to be strongly related to poor overall survival disease-free survival. This indicated that there was a positive correlation between poor prognostic-related AS events and SFs. The SRSF2 gene encodes for the serine/arginine rich family of pre-mRNA SFs, and they constitute part of the spliceosome and are critical in mRNA spicing [35]. These SFs have an RNA recognition motif that binds RNA and an RS motif that bind other proteins. SRSF2 acts as a splicing activator; it stabilizes the RNA and takes part in translation. Mutation in SRSF2 is associated with worse prognosis in hematopoietic disease [36]. The findings of the present study also suggest that several poor prognosis-related AS events were highly related to high levels of tumor-promoting SFs.
Conclusionss
The prognosis of HCC patients based on aberrant variation in AS and SFs was comprehensively evaluated. Although the predictive value of AS events was not validated, this study demonstrates that AS and SFs are potential therapeutic targets and biomarkers of HCC. However, more effective AS-based genome-wide analyses are needed for better prognosis prediction and identification of therapeutic biomarkers of HCC. Indeed, this and other studies advance the prognosis prediction of HCC and the design of novel therapeutic strategies.
Figures
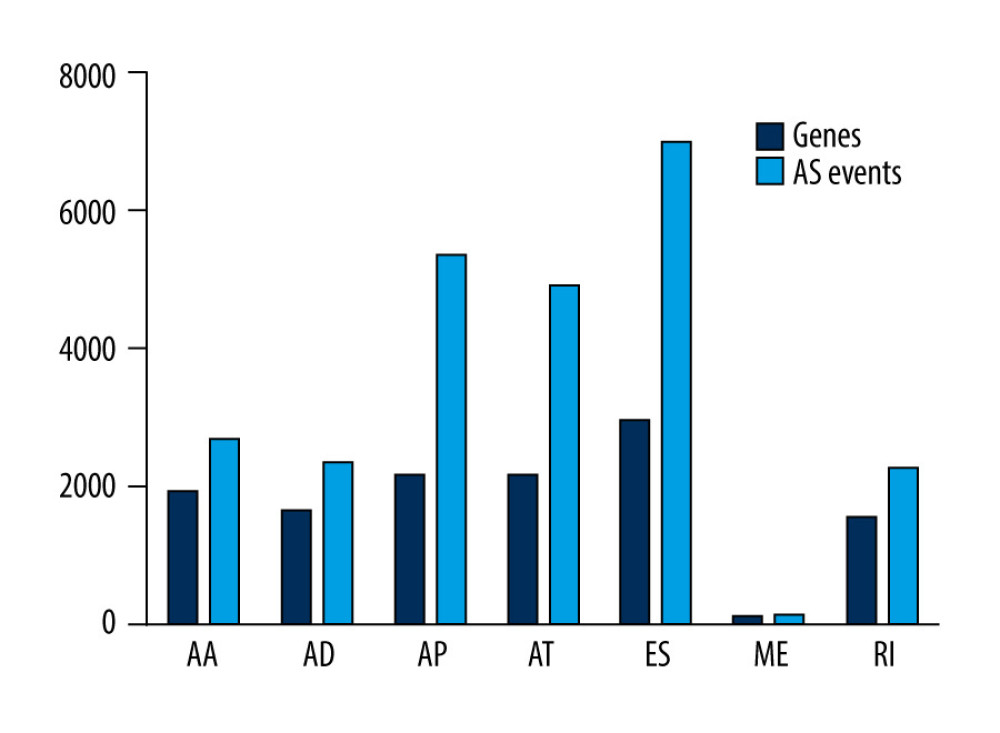 Figure 1. AS events and corresponding genes in this study.
Figure 1. AS events and corresponding genes in this study. 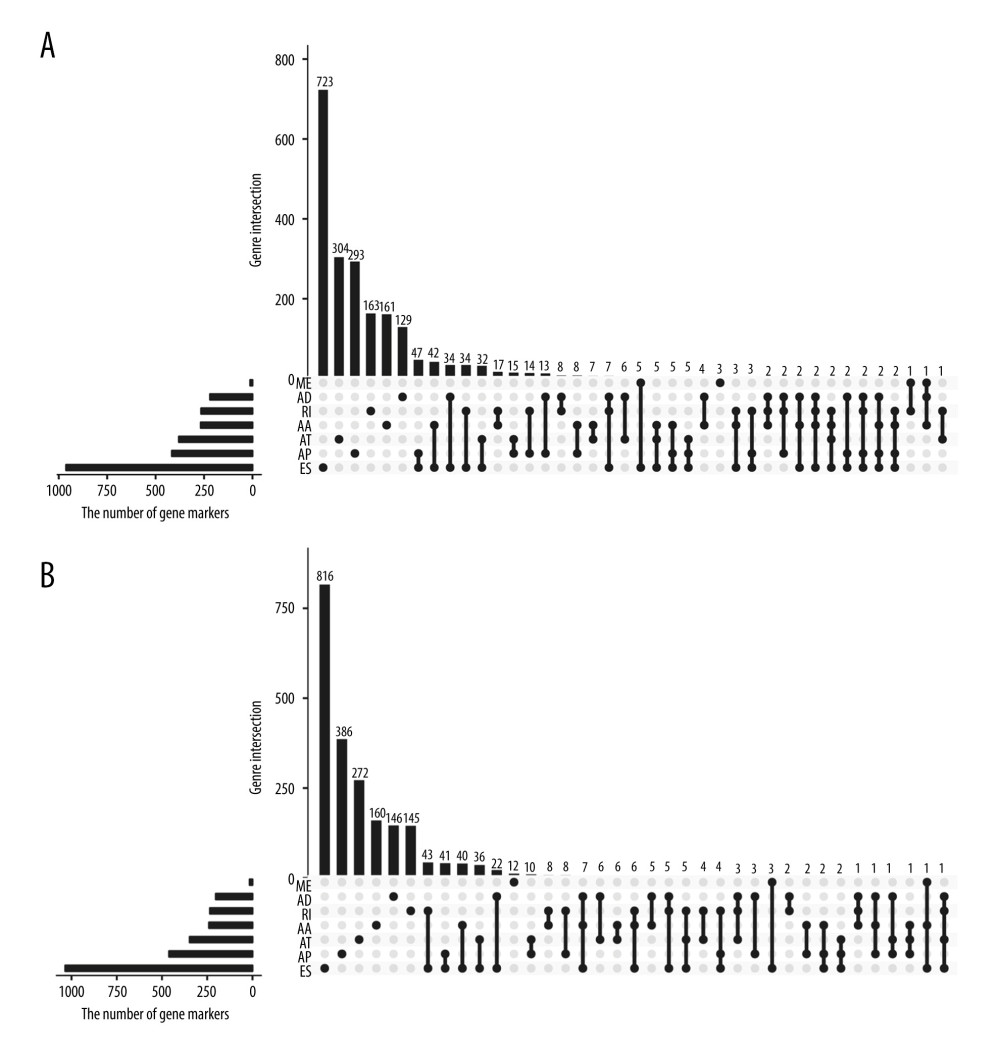 Figure 2. Prognosis-related AS events in HCC patients. (A) UpSet plot of interactions among the 7 types of recurrence-free survival-associated AS events. (B) UpSet plot showing interactions among the 7 types of AS events associated overall survival.
Figure 2. Prognosis-related AS events in HCC patients. (A) UpSet plot of interactions among the 7 types of recurrence-free survival-associated AS events. (B) UpSet plot showing interactions among the 7 types of AS events associated overall survival.  Figure 3. Establishment of prognostic signatures based on LASSO Cox analysis. The coefficients obtained from the LASSO algorithm: (A) AA, (B) DA, (C) AP, (D) AT, (E) ES, (F) RI, and (G) ALL AS types.
Figure 3. Establishment of prognostic signatures based on LASSO Cox analysis. The coefficients obtained from the LASSO algorithm: (A) AA, (B) DA, (C) AP, (D) AT, (E) ES, (F) RI, and (G) ALL AS types.  Figure 4. The partial likelihood deviance obtained from the LASSO algorithm: (A) AA, (B) DA, (C) AP, (D) AT, (E) ES, (F) RI, and (G) ALL AS types.
Figure 4. The partial likelihood deviance obtained from the LASSO algorithm: (A) AA, (B) DA, (C) AP, (D) AT, (E) ES, (F) RI, and (G) ALL AS types. 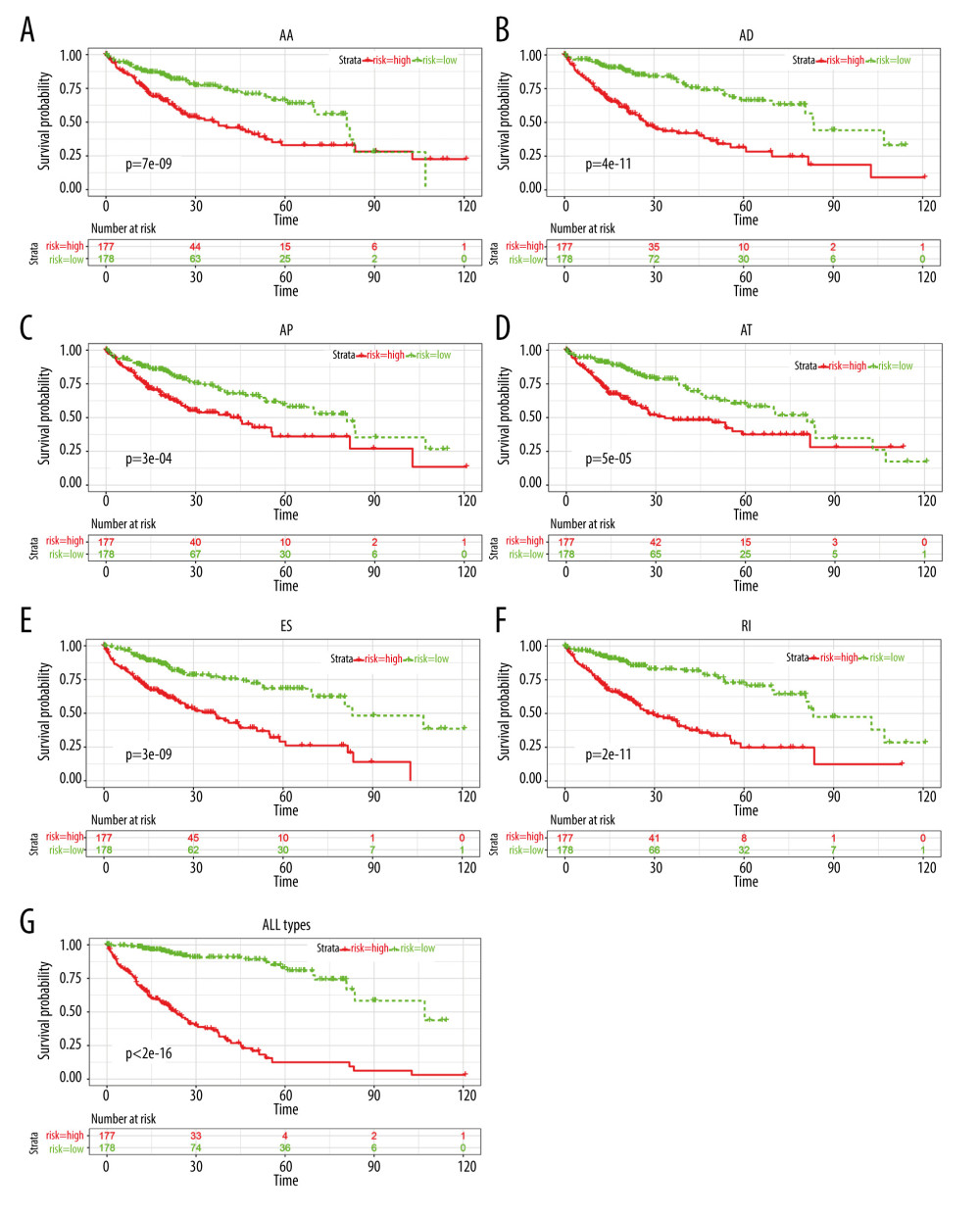 Figure 5. Kaplan-Meier plot of prognostic predictors in HCC patients. (A–F) Kaplan-Meier plot showing the overall survival probability over time for prognosis prediction of 6 types of AS events with low (green) risk group and high (red). (G) Kaplan-Meier plot showing the survival probability over time for the final prognostic predictor with low-risk group (green) and high-risk group (red).
Figure 5. Kaplan-Meier plot of prognostic predictors in HCC patients. (A–F) Kaplan-Meier plot showing the overall survival probability over time for prognosis prediction of 6 types of AS events with low (green) risk group and high (red). (G) Kaplan-Meier plot showing the survival probability over time for the final prognostic predictor with low-risk group (green) and high-risk group (red). 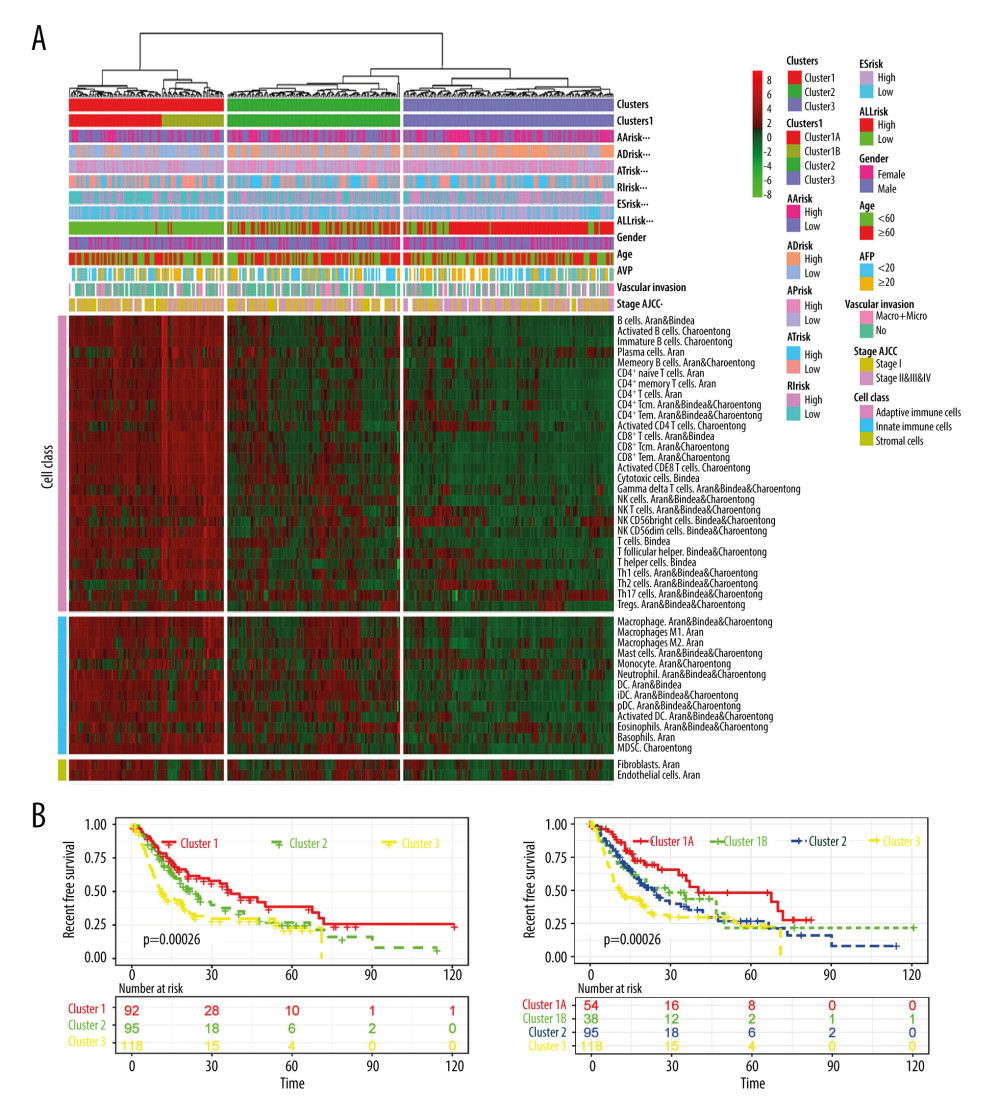 Figure 6. The relationship between AS events and the tumor immune microenvironment. (A) Clustering of HCC microenvironment phenotypes based on the estimated scores of 43 cell subsets. The average score of the cell type was used as indicated by Aran, Bindea, or Charoentong in cases where there were common cell types among the different methods (Aran, Bindea, and Charoentong). (B) Recurrence-free survival analyses for the different immune subtypes; left panel: 3 immune clusters and right panel: cluster1 divided into active (cluster 1A) and exhausted (cluster 1B) subtypes.
Figure 6. The relationship between AS events and the tumor immune microenvironment. (A) Clustering of HCC microenvironment phenotypes based on the estimated scores of 43 cell subsets. The average score of the cell type was used as indicated by Aran, Bindea, or Charoentong in cases where there were common cell types among the different methods (Aran, Bindea, and Charoentong). (B) Recurrence-free survival analyses for the different immune subtypes; left panel: 3 immune clusters and right panel: cluster1 divided into active (cluster 1A) and exhausted (cluster 1B) subtypes. 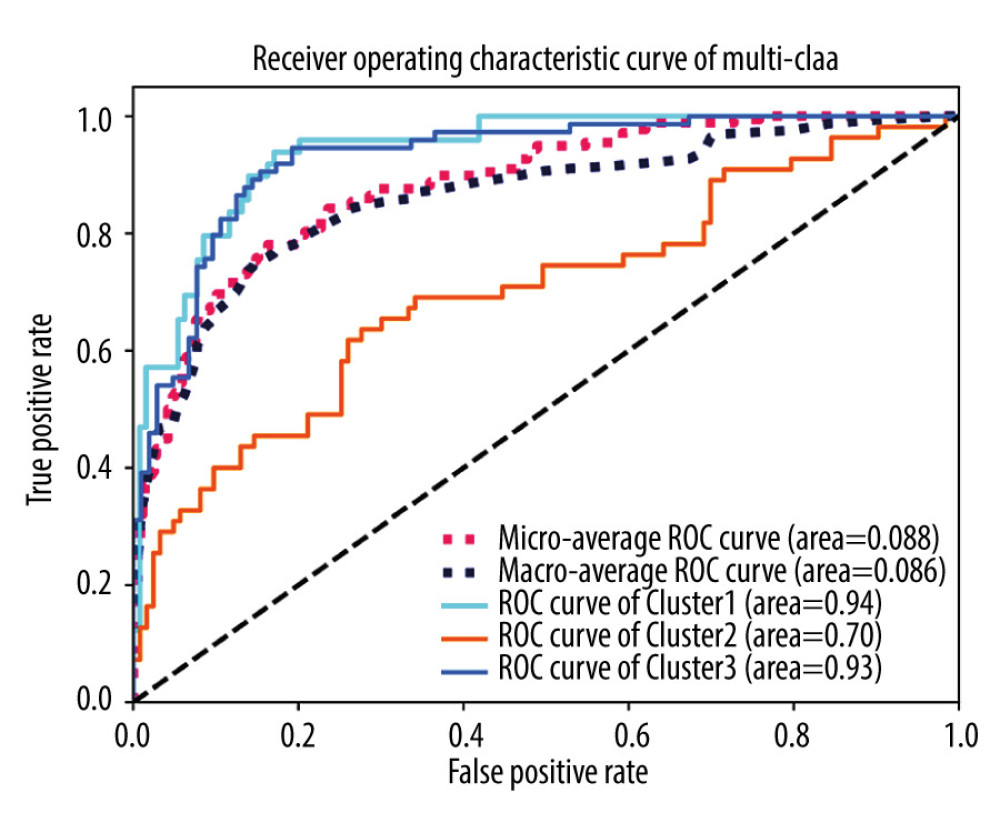 Figure 7. ROC curves of immune clusters predicted by AS events. Micro and macro methods for average ROC were used.
Figure 7. ROC curves of immune clusters predicted by AS events. Micro and macro methods for average ROC were used. 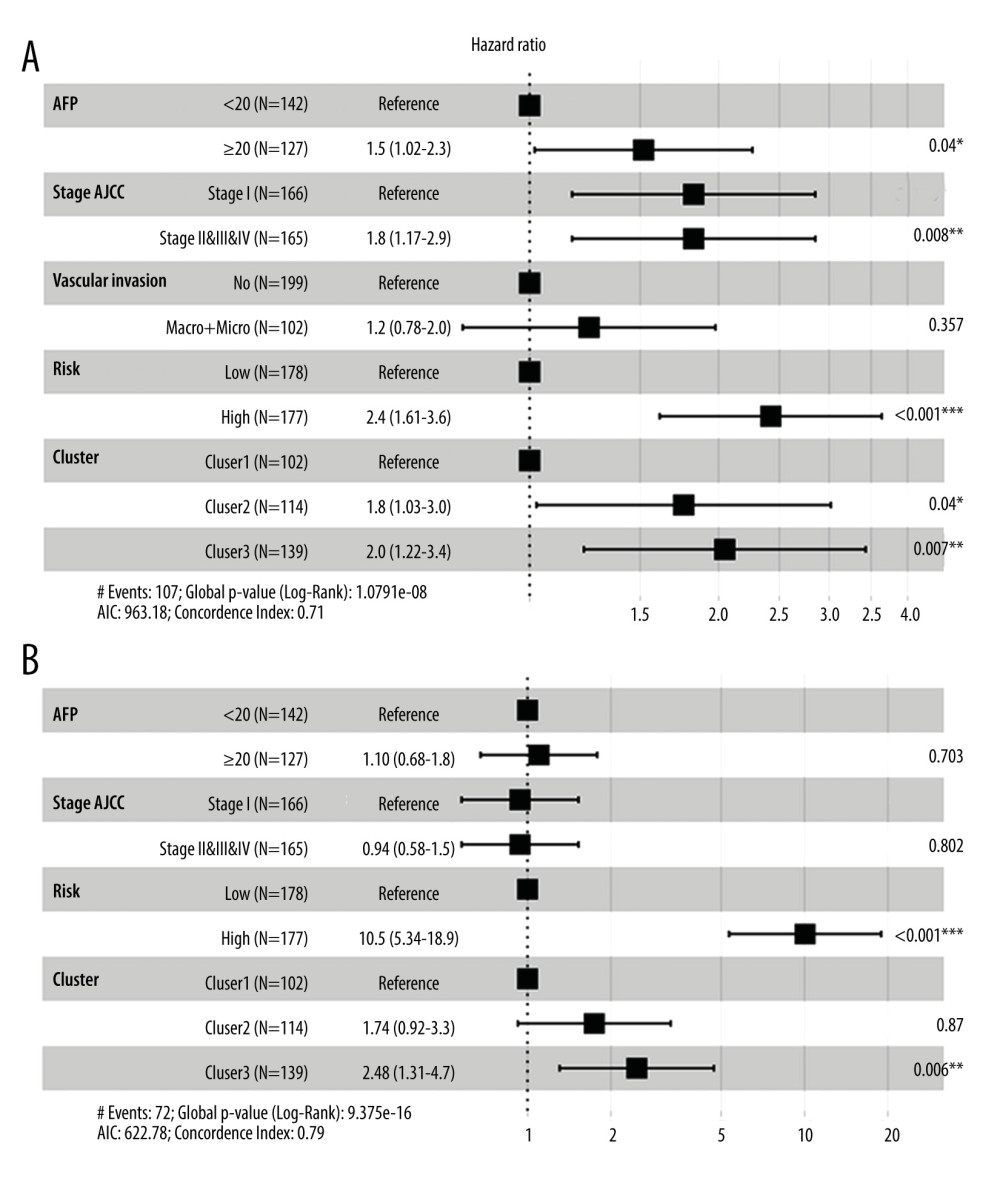 Figure 8. Multivariate analyses of the association between clinical factors, immune clusters and the risk score for recurrence-free survival (A) and overall survival (B) of HCC patients. The data are presented as hazard ratios (HR) with 95% confidence intervals.
Figure 8. Multivariate analyses of the association between clinical factors, immune clusters and the risk score for recurrence-free survival (A) and overall survival (B) of HCC patients. The data are presented as hazard ratios (HR) with 95% confidence intervals. 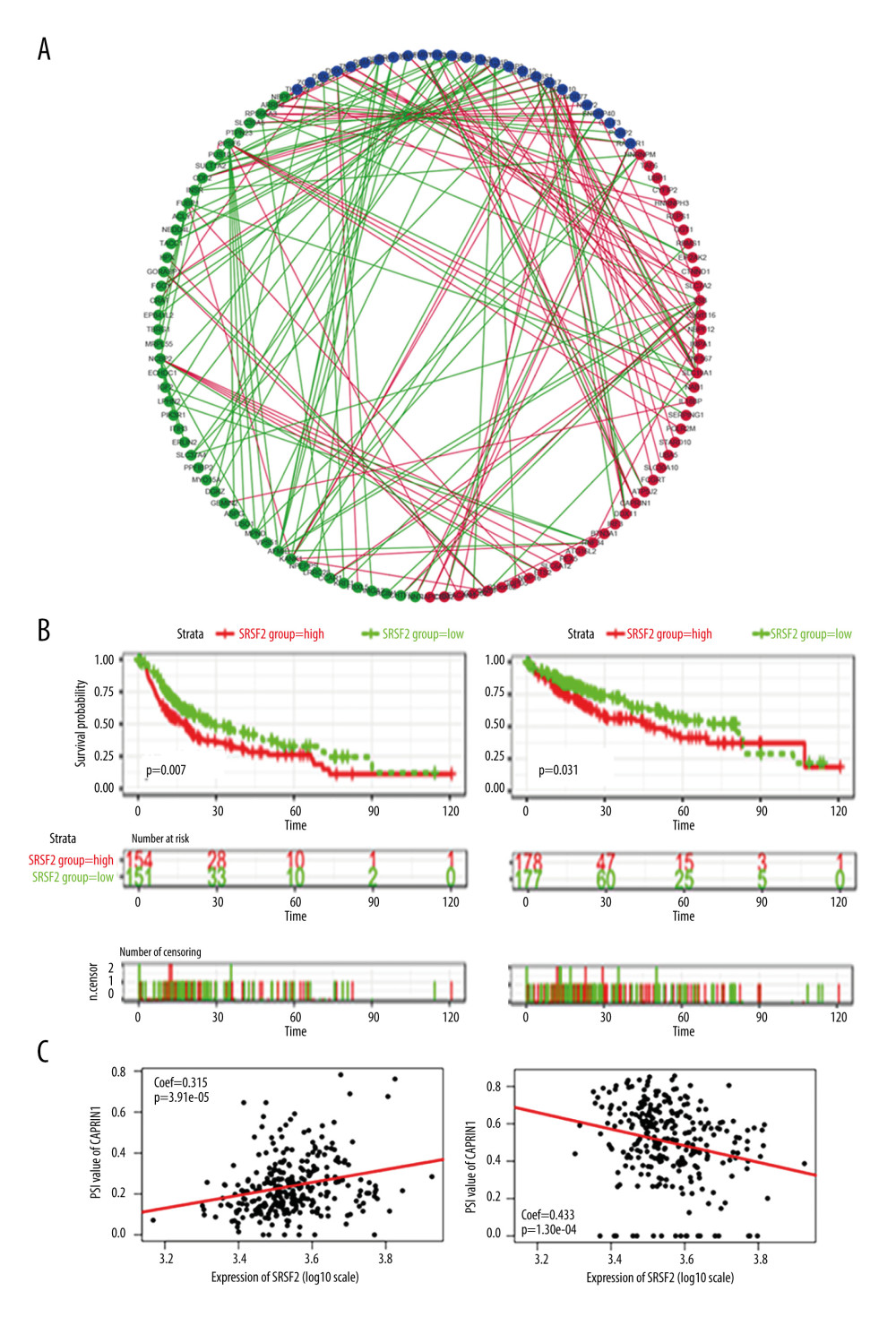 Figure 9. Survival-associated SFs and splicing correlation network in HCC patients. (A) Splicing correlation network. AS events with PSI values negatively/positively associated with survival are shown in red/green dots, respectively. SFs related to survival are presented as purple dots. Green/red lines represent negative/positive between SFs expression and PSI values of AS, respectively. (B) High expression (red line) of splicing factor SRSF2 is strongly related to poor disease-free survival (left panel) and poor overall survival (right panel). (C) Dot plot of correlation between expression of SRSF2 and AP PSI values of CAPRIN1 (left panel), and the correlation between ES PSI values of SULT1A2 and expression of SRSF2 (right panel).
Figure 9. Survival-associated SFs and splicing correlation network in HCC patients. (A) Splicing correlation network. AS events with PSI values negatively/positively associated with survival are shown in red/green dots, respectively. SFs related to survival are presented as purple dots. Green/red lines represent negative/positive between SFs expression and PSI values of AS, respectively. (B) High expression (red line) of splicing factor SRSF2 is strongly related to poor disease-free survival (left panel) and poor overall survival (right panel). (C) Dot plot of correlation between expression of SRSF2 and AP PSI values of CAPRIN1 (left panel), and the correlation between ES PSI values of SULT1A2 and expression of SRSF2 (right panel). Tables
Supplementary Table 1. Sets of gene and splice event for each type of AS associated with OS.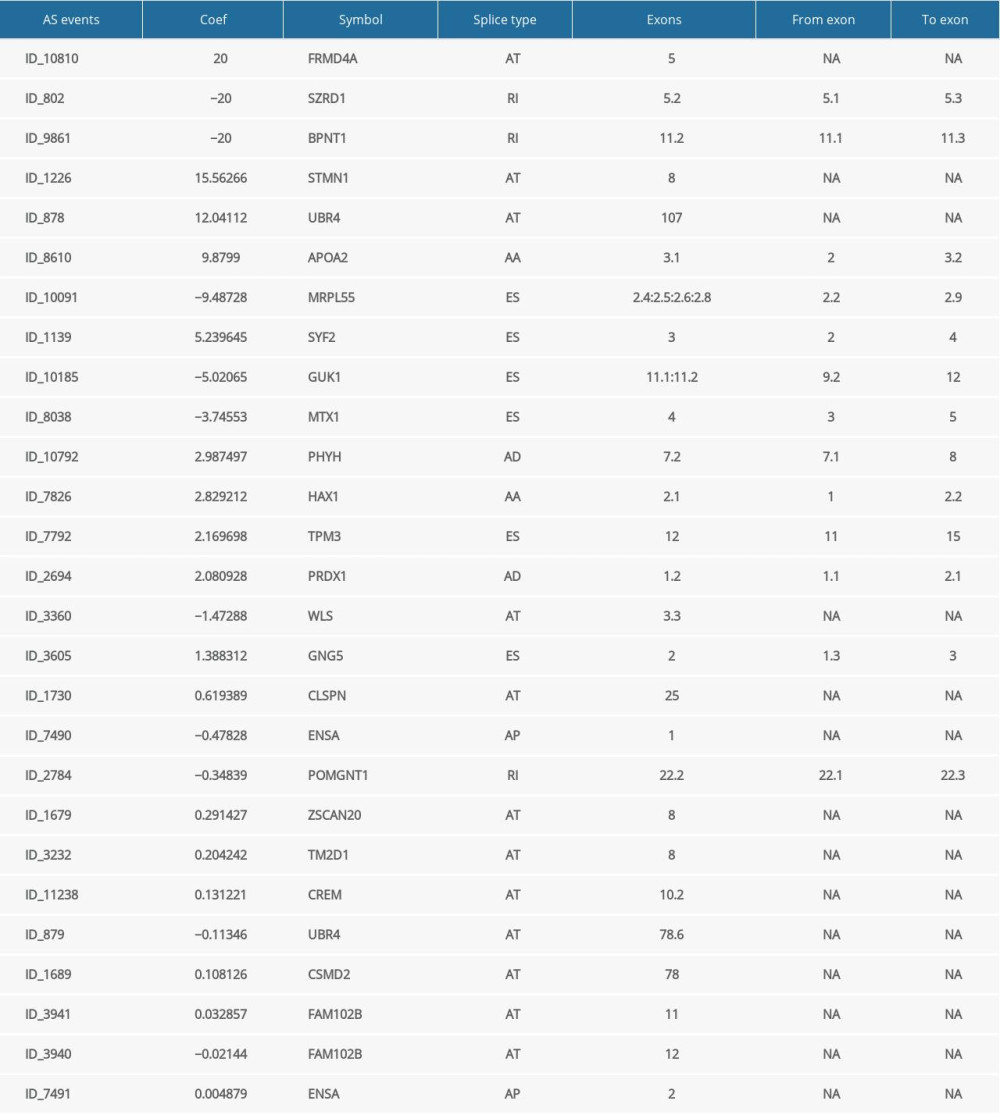 Supplementary Table 3. Univariate Cox analysis of clinical characteristics, the risk score (calculated from the final prognostic signatures) and immune clusters on RFS.
Supplementary Table 3. Univariate Cox analysis of clinical characteristics, the risk score (calculated from the final prognostic signatures) and immune clusters on RFS.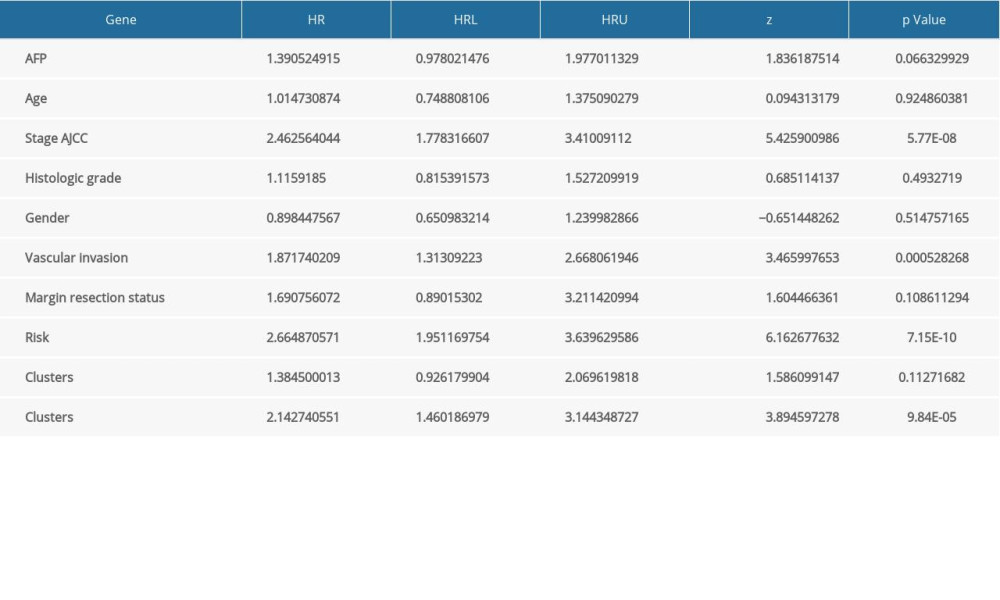 Supplementary Table 4. Univariate Cox analysis of clinical characteristics, the risk score (calculated from the final prognostic signatures), and immune clusters on OS.
Supplementary Table 4. Univariate Cox analysis of clinical characteristics, the risk score (calculated from the final prognostic signatures), and immune clusters on OS.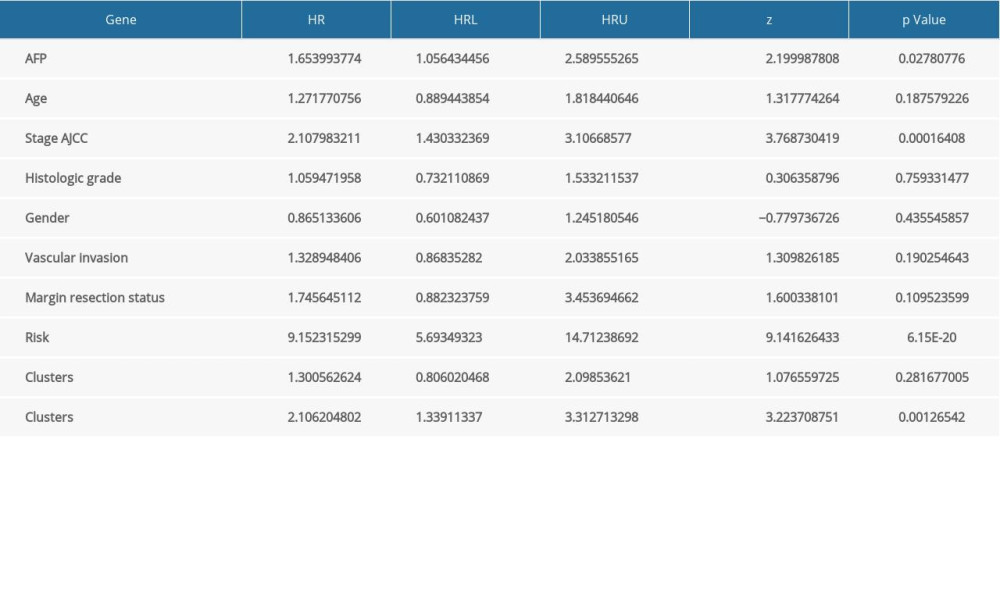
References
1. Nilsen TW, Graveley BR, Expansion of the eukaryotic proteome by alternative splicing: Nature, 2010; 463(7280); 457-63
2. Salton M, Misteli T, Small molecule modulators of pre-mRNA splicing in cancer therapy: Trends Mol Med, 2016; 22(1); 28-37
3. Lin P, He R-Q, Huang Z-G, Role of global aberrant alternative splicing events in papillary thyroid cancer prognosis: Aging, 2019; 11(7); 2082-97
4. Koh CM, Bezzi M, Low DHP, MYC regulates the core pre-mRNA splicing machinery as an essential step in lymphomagenesis: Nature, 2015; 523(7558); 96-100
5. Kim E, Goren A, Ast G, Insights into the connection between cancer and alternative splicing: Trends Genet, 2008; 24(1); 7-10
6. Oltean S, Bates DO, Hallmarks of alternative splicing in cancer: Oncogene, 2014; 33(46); 5311-18
7. Teng H, Mao F, Liang J, Transcriptomic signature associated with carcinogenesis and aggressiveness of papillary thyroid carcinoma: Theranostics, 2018; 8(16); 4345-58
8. Llovet JM, Zucman-Rossi J, Pikarsky E, Hepatocellular carcinoma: Nat Rev Dis Primers, 2016; 2(1); 16018
9. Chen Y, ECY , Gong ZW, Chimeric antigen receptor-engineered T-cell therapy for liver cancer: Hepatobiliary Pancreat Dis Int, 2018; 17(4); 301-9
10. Voutsadakis IA, PD-1 inhibitors monotherapy in hepatocellular carcinoma: Meta-analysis and systematic review: Hepatobiliary Pancreat Dis Int, 2019; 18(6); 505-10
11. Rigden DJ, Fernández-Suárez XM, Galperin MY, The 2016 database issue of Nucleic Acids Research and an updated molecular biology database collection: Nucleic Acids Res, 2016; 44(D1); D1-D6
12. Lex A, Gehlenborg N, Strobelt H, UpSet: Visualization of intersecting sets: IEEE Trans Vis Comput Graph, 2014; 20(12); 1983-92
13. Tsai YS, Dominguez D, Gomez SM, Wang Z, Transcriptome-wide identification and study of cancer-specific splicing events across multiple tumors: Oncotarget, 2015; 6(9); 6825-39
14. Kim HK, Pham MHC, Ko KS, Alternative splicing isoforms in health and disease: Pflügers Arch, 2018; 470(7); 995-1016
15. Tremblay MP, Armero VE, Allaire A, Global profiling of alternative RNA splicing events provides insights into molecular differences between various types of hepatocellular carcinoma: BMC Genomics, 2016; 17(1); 683
16. Li S, Hu Z, Zhao Y, Huang S, He X, Transcriptome-wide analysis reveals the landscape of aberrant alternative splicing events in liver cancer: Hepatology, 2019; 69(1); 359-75
17. Xiong Y, Yang G, Wang K, Genome-wide transcriptional analysis reveals alternative splicing event profiles in hepatocellular carcinoma and their prognostic significance: Front Genet, 2020; 11; 879
18. Zhang D, Duan Y, Wang Z, Lin J, Systematic profiling of a novel prognostic alternative splicing signature in hepatocellular carcinoma: Oncol Rep, 2019; 42(6); 2450-72
19. Liu J, Lichtenberg T, Hoadley KA, An integrated TCGA pan-cancer clinical data resource to drive high-quality survival outcome analytics: Cell, 2018; 173(2); 400-416e411
20. Ryan M, Wong WC, Brown R, TCGASpliceSeq a compendium of alternative mRNA splicing in cancer: Nucleic Acids Res, 2016; 44(D1); D1018-22
21. Ryan MC, Cleland J, Kim R, SpliceSeq: A resource for analysis and visualization of RNA-Seq data on alternative splicing and its functional impacts: Bioinformatics, 2012; 28(18); 2385-87
22. Tibshirani R, The lasso method for variable selection in the Cox model: Stat Med, 1997; 16(4); 385-95
23. Charoentong P, Finotello F, Angelova M, Pan-cancer immunogenomic analyses reveal genotype-immunophenotype relationships and predictors of response to checkpoint blockade: Cell Rep, 2017; 18(1); 248-62
24. Aran D, Hu Z, Butte AJ, xCell: Digitally portraying the tissue cellular heterogeneity landscape: Genome Biol, 2017; 18(1); 220
25. Bindea G, Mlecnik B, Tosolini M, Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer: Immunity, 2013; 39(4); 782-95
26. Shen S, Wang Y, Wang C, SURVIV for survival analysis of mRNA isoform variation: Nat Commun, 2016; 7(1); 11548
27. Liu S, Cheng C, Alternative RNA splicing and cancer: WIREs RNA, 2013; 4(5); 547-66
28. Zeilstra J, Joosten SPJ, van Andel H, Stem cell CD44v isoforms promote intestinal cancer formation in Apc(min) mice downstream of Wnt signaling: Oncogene, 2014; 33(5); 665-70
29. Li Y, Sun N, Lu Z, Sun S, Prognostic alternative mRNA splicing signature in non-small cell lung cancer: Cancer Lett, 2017; 393; 40-51
30. Lin P, He RQ, Huang ZG, Role of global aberrant alternative splicing events in papillary thyroid cancer prognosis: Aging, 2019; 11(7); 2082-97
31. Wang L, Yao M, Fang M, Expression of hepatic Wnt5a and its clinicopathological features in patients with hepatocellular carcinoma: Hepatobiliary Pancreat Dis Int, 2018; 17(3); 227-32
32. Behne T, Copur MS, Biomarkers for hepatocellular carcinoma: Int J Hepatol, 2012; 2012; 859076
33. Charenton C, Wilkinson ME, Nagai K, Mechanism of 5′ splice site transfer for human spliceosome activation: Science, 2019; 364(6438); 362-67
34. Hong W, Zhang W, Guan R, Genome-wide profiling of prognosis-related alternative splicing signatures in sarcoma: Anna Transl Med, 2019; 7(20); 557
35. Pandit S, Zhou Y, Shiue L, Genome-wide analysis reveals SR protein cooperation and competition in regulated splicing: Mol Cell, 2013; 50(2); 223-35
36. Zhang J, Lieu YK, Ali AM, Disease-associated mutation in SRSF2 misregulates splicing by altering RNA-binding affinities: Proc Natl Acad Sci USA, 2015; 112(34); E4726-34
Figures
Figure 1. AS events and corresponding genes in this study.Figure 2. Prognosis-related AS events in HCC patients. (A) UpSet plot of interactions among the 7 types of recurrence-free survival-associated AS events. (B) UpSet plot showing interactions among the 7 types of AS events associated overall survival.Figure 3. Establishment of prognostic signatures based on LASSO Cox analysis. The coefficients obtained from the LASSO algorithm: (A) AA, (B) DA, (C) AP, (D) AT, (E) ES, (F) RI, and (G) ALL AS types.Figure 4. The partial likelihood deviance obtained from the LASSO algorithm: (A) AA, (B) DA, (C) AP, (D) AT, (E) ES, (F) RI, and (G) ALL AS types.Figure 5. Kaplan-Meier plot of prognostic predictors in HCC patients. (A–F) Kaplan-Meier plot showing the overall survival probability over time for prognosis prediction of 6 types of AS events with low (green) risk group and high (red). (G) Kaplan-Meier plot showing the survival probability over time for the final prognostic predictor with low-risk group (green) and high-risk group (red).Figure 6. The relationship between AS events and the tumor immune microenvironment. (A) Clustering of HCC microenvironment phenotypes based on the estimated scores of 43 cell subsets. The average score of the cell type was used as indicated by Aran, Bindea, or Charoentong in cases where there were common cell types among the different methods (Aran, Bindea, and Charoentong). (B) Recurrence-free survival analyses for the different immune subtypes; left panel: 3 immune clusters and right panel: cluster1 divided into active (cluster 1A) and exhausted (cluster 1B) subtypes.Figure 7. ROC curves of immune clusters predicted by AS events. Micro and macro methods for average ROC were used.Figure 8. Multivariate analyses of the association between clinical factors, immune clusters and the risk score for recurrence-free survival (A) and overall survival (B) of HCC patients. The data are presented as hazard ratios (HR) with 95% confidence intervals.Figure 9. Survival-associated SFs and splicing correlation network in HCC patients. (A) Splicing correlation network. AS events with PSI values negatively/positively associated with survival are shown in red/green dots, respectively. SFs related to survival are presented as purple dots. Green/red lines represent negative/positive between SFs expression and PSI values of AS, respectively. (B) High expression (red line) of splicing factor SRSF2 is strongly related to poor disease-free survival (left panel) and poor overall survival (right panel). (C) Dot plot of correlation between expression of SRSF2 and AP PSI values of CAPRIN1 (left panel), and the correlation between ES PSI values of SULT1A2 and expression of SRSF2 (right panel). Tables
Supplementary Table 1. Sets of gene and splice event for each type of AS associated with OS.Supplementary Table 3. Univariate Cox analysis of clinical characteristics, the risk score (calculated from the final prognostic signatures) and immune clusters on RFS.Supplementary Table 4. Univariate Cox analysis of clinical characteristics, the risk score (calculated from the final prognostic signatures), and immune clusters on OS. In Press
Clinical Research
Effects of Single-Bout Endurance Exercise Intensity on Peripheral Neurotrophic Factors in Patients With Isc...Med Sci Monit In Press; DOI: 10.12659/MSM.952089
Review article
Anisodus tanguticus in Cancer Research: A Review of Traditional Use, Phytochemistry, Extraction Methods, an...Med Sci Monit In Press; DOI: 10.12659/MSM.952999
Clinical Research
Nasal Mucociliary Clearance and Its Relationship With Disease Severity in Patients With Multiple SclerosisMed Sci Monit In Press; DOI: 10.12659/MSM.952850
Clinical Research
Modified Thoracoabdominal Nerves Block Through the Perichondrial Approach vs Subcostal Transversus Abdomini...Med Sci Monit In Press; DOI: 10.12659/MSM.953976
Most Viewed Current Articles
17 Jan 2024 : Review article 14,176,570
Vaccination Guidelines for Pregnant Women: Addressing COVID-19 and the Omicron VariantDOI :10.12659/MSM.942799
Med Sci Monit 2024; 30:e942799
13 Nov 2021 : Clinical Research 3,762,188
Acceptance of COVID-19 Vaccination and Its Associated Factors Among Cancer Patients Attending the Oncology ...DOI :10.12659/MSM.932788
Med Sci Monit 2021; 27:e932788
14 Dec 2022 : Clinical Research 2,466,310
Prevalence and Variability of Allergen-Specific Immunoglobulin E in Patients with Elevated Tryptase LevelsDOI :10.12659/MSM.937990
Med Sci Monit 2022; 28:e937990
16 May 2023 : Clinical Research 708,927
Electrophysiological Testing for an Auditory Processing Disorder and Reading Performance in 54 School Stude...DOI :10.12659/MSM.940387
Med Sci Monit 2023; 29:e940387