03 April 2021: Animal Study
Expression of DUSP12 Reduces Lung Vascular Endothelial Cell Damage in a Murine Model of Lipopolysaccharide-Induced Acute Lung Injury via the Apoptosis Signal-Regulating Kinase 1 (ASK1)-Jun N-Terminal Kinase Activation (JNK) Pathway
Zhao Hui1BF, Huang Jie2CD, Guo-Hua Fan2ABCDEFG*DOI: 10.12659/MSM.930429
Med Sci Monit 2021; 27:e930429






Abstract
BACKGROUND: Acute lung injury (ALI) results from damage to the alveolar capillary endothelial cells and can result in acute respiratory distress syndrome (ARDS). This study aimed to investigate murine lung vascular endothelial cells (MLECs) damage in a murine model of lipopolysaccharide (LPS)-induced ALI.
MATERIAL AND METHODS: Mice were injected with LPS to induce an acute lung injury model. An adenovirus transfection system was used to overexpress or knockdown DUSP12 in mice. MLECs were isolated, cultured and transfected with DUSP12-overexpressing adenovirus or with DUSP12 siRNA to knockdown DUSP12. LPS was used to establish a cell injury model. ELISA and RT-PCR were used to examine cell inflammation. LPS-induced oxidative stress was also evaluated using commercial kits.
RESULTS: A decreased level of DUSP12 was observed in MLECs treated with LPS. DUSP12 overexpression in mice attenuated LPS-induced lung inflammation and lung injury, as reflected by reduced levels of proinflammatory cytokines. Mice with DUSP12 knockdown exhibited worsened lung inflammation and injury. In vitro, DUSP12 overexpression in endothelial cells ameliorated LPS-induced inflammation, apoptosis, and oxidative stress. DUSP12 silencing in endothelial cells aggravated LPS-induced inflammation, apoptosis, and oxidative stress. Furthermore, we found that DUSP12 directly bound to apoptosis signal-regulating kinase 1 (ASK1) to inhibit Jun N-terminal kinase activation (JNK). A JNK1/2 inhibitor and ASK1 siRNA ameliorated the exacerbating effects of DUSP12 knockdown in vitro.
CONCLUSIONS: Our data demonstrated that DUSP12 suppressed MLEC injury in response to LPS insult by regulating the ASK1/JNK pathway.
Keywords: acute lung injury, Dual-Specificity Phosphatases, endothelial cells, JNK Mitogen-Activated Protein Kinases, Lipopolysaccharides, MAP Kinase Kinase 4, MAP Kinase Kinase Kinase 5
Background
Acute lung injury (ALI) induced by sepsis, bacterial infections, viral pneumonia, and trauma is a major cause of acute respiratory distress syndrome (ARDS), which is characterized by severe hypoxemia and bilateral lung osmotic injury [1]. During the development of ARDS, damage to alveolar capillary endothelial cells leads to increased barrier permeability, causes a large amount of edema and inflammatory fluid infiltration into the alveolar cavity, weakens gas exchange, and eventually leads to respiratory failure [2,3]. Although the application of a lung-protective ventilation strategy has reduced the mortality of ALI, there is currently no clinically effective drug to treat ALI. Thus, deeply understanding the pathology of ALI and seeking new therapeutic targets are of great importance.
The bispecific phosphatase family is a family of proteins that can dephosphorylate tyrosine and serine/threonine. DUSP12 is a member of the dual-specificity phosphatase (DUSP) subfamily of protein-tyrosine phosphatases (PTPs). DUSP12 contains a catalytic domain, a zinc finger domain, and an aldehyde dehydrogenase active site [4]. DUSP12 is widely expressed in a variety of tissues and organs, including the lungs [5]. Since researchers discovered DUSP12 in 2005, studies have shown that DUSP12 is involved in the regulation of apoptosis and proliferation [6]. Many bispecific phosphatases dephosphorylate mitogen-activated protein kinases (MAPKs) and phosphatidylinositol 3-kinases (PI3Ks), which function in many inflammatory and apoptosis-associated diseases [4,7]. A previous study reported that hDUSP12 could regulate cellular DNA levels [8]. hDUSP12 gene polymorphisms are associated with glucose metabolism in the Chinese population [9]. hDUSP12 has also been reported to be associated with multiple ribonucleoproteins [10]. Whether DUSP12 is involved in ALI is unclear. This study aimed to investigate murine lung vascular endothelial cell (MLVEC) damage in a murine model of lipopolysaccharide (LPS)-induced acute lung injury (ALI).
Material and Methods
ANIMALS:
Male C57BL/6J mice aged 8–10 weeks (weighing 23.5–27.5 g) were purchased from the Chinese Academy of Medical Sciences and Peking Union Medical College. The animal experiments were performed according to the Guide for the Care and Use of Laboratory Animals published by the U.S. National Institutes of Health (NIH Publication No. 85-23, revised 1996) and were approved by the Animal Care and Use Committee of our hospital.
MOUSE MODEL:
LPS was used to establish an acute lung injury model. Briefly, after anesthetization with 3% amobarbital, LPS was injected into the mice (100 μg diluted in 20 μL of sterile normal saline, injected into the trachea and incubated for 48 h). The control mice were administered the same volume of normal saline. To overexpress or knock down DUSP12, the mice were also injected with Ad-DUSP12 or Ad-shDUSP12 6 h after LPS injection (each with 60 μl, 5.0–6.5×1013 GC/ml, caudal vein. Forty-eight hours after LPS injection, the mice were sacrificed, the lungs were washed with PBS 3 times, and the solution was collected. To inhibit JNK, the mice were administered SP600125 by intraperitoneal injection (15 mg/kg, Sigma-Aldrich, USA) at the same time as LPS administration.
HISTOLOGICAL ANALYSIS:
Forty-eight hours after LPS injection, the right lung tissue was collected and fixed in 10% formalin. After being embedded in paraffin, the lung tissues were stained with hematoxylin and eosin (H&E) to detect inflammation and determine lung injury scores by using a digital camera (Olympus BX 53 microscope, Tokyo, Japan). Lung tissues were also immunofluorescently stained for CD31 and DUSP12 to detect the expression of DUSP12 after Ad-DUSP12 or Ad-shDUSP12 injection.
DETERMINATION OF PULMONARY EDEMA:
Forty-eight hours after LPS injection, the right lung tissue was collected and weighed to determine the wet weight. Then, the lung tissues were baked at 80°C for 48 h to determine the dry weight. Then, the W/D weight ratio was calculated.
LUNG INJURY SCORE:
The lung injury score was calculated according to the criteria described by the official American Thoracic Society workshop report on the features and measurements of experimental acute lung injury in animals as follows:
The lung injury score was calculated as [(20×A)+(14×B)+ (7×C)+(7×D)+(2×E)]/(number of fields×100) [11].
CELL CULTURE AND TREATMENT:
We isolated murine lung vascular endothelial cells (MLECs) according to a previous protocol [12]. Cells were cultured in DMEM containing glucose, 20% fetal bovine serum, 1% MEM nonessential amino acids, 2.5% HEPES buffer solution (1 M) (Gibco, USA), and endothelial cell growth supplement (0.1 mg/ml) (Sigma, USA). To overexpress DUSP12, cells were transfected with Ad-DUSP12 (MOI=75) for 6 h and then stimulated with LPS (100 μg/ml) for 24 h. Cells were transfected with DUSP12 siRNA and negative control scRNA (RiboBio, Shanghai, China) in the presence of Lipo2000™ transfection reagent (Beyotime, Shanghai, China). To inhibit JNK1/2, cells were treated with SP600125 (10 μM, MedChemExpress). To silence ASK1, cells were transfected with ASK1 siRNA (RiboBio, Shanghai, China).
ELISA:
ELISA kits were used to measure TNF-α, IL-1 and IL-6 levels in bronchoalveolar lavage fluid from ALI mice and cell lysates according to the manufacturer’s protocol. An ELISA plate reader (Synergy HT, Biotek, Vermont, USA) was used to measure the optical density at 450 nm.
OXIDATIVE STRESS ANALYSIS:
The reactive oxygen species (ROS) level was measured by using a 2′,7′-dichlorofluorescin diacetate (DCFH-DA) probe according to the manufacturer’s protocol. Commercial kits (Beyotime, Shanghai, China) were used to detect superoxide dismutase 2, mitochondrial (SOD2), glutathione peroxidase (Gpx), nicotinamide adenine dinucleotide phosphate (NADPH) activity, malondialdehyde (MDA) levels, and caspase3 activity.
TUNEL STAINING:
Terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) staining was performed to detect cell apoptosis. Briefly, 4% paraformaldehyde was used to fix the cells. Triton™ X-100 (0.1%) was used to permeabilize the cells for 5 min. Then, the cells were stained with the TUNEL assay kit for 1 h. DAPI was used to stain the nuclei. A fluorescence microscope (Olympus DX51) was used to capture images.
RT-PCR AND WESTERN BLOTTING:
For total mRNA isolation, lung tissue or cells were lysed in TRIzol reagent. mRNA purity was determined by using OD260/OD280 ratios measured with a SmartSpec Plus Spectrophotometer (Bio-Rad). A cDNA synthesis kit (Roche Diagnostics) was used to reverse transcribe 2 μg of mRNA into cDNA. A LightCycler 480 SYBR Green I Master kit (Roche Diagnostics) was used for amplification. The glyceraldehyde 3-phosphate dehydrogenase (GAPDH) gene was used as a reference.
For western blotting, radioimmunoprecipitation (RIPA) lysis buffer was used to lyse the cells. The BCA method was used to determine protein concentrations. Proteins were then loaded onto 10% SDS-PAGE gels and transferred to polyvinylidene difluoride (PVDF) membranes (Millipore). After being incubated with primary antibodies at 4°C overnight, the blots were color-rendered with enhanced chemiluminescence (ECL) reagents (Bio-Rad, Hercules, CA, USA) and scanned by a ChemiDoc MP imaging system (Bio-Rad). GAPDH was used as a reference protein.
COIMMUNOPRECIPITATION ASSAYS:
MLECs were transfected with psicoR-HA-DUSP12 and psicoRFlag-ASK1. Then, the cells were lysed with immunoprecipitation buffer. One microgram of antibody and 10 μL of protein A/G-agarose beads were added to the immunoprecipitate and incubated overnight at 4°C. Then, we used immunoprecipitation buffer to wash the beads. After the proteins were collected, the samples were immunoblotted with the indicated antibodies.
DATA ANALYSIS:
We used SPSS 23.3 to analysis the data. The
Results
DUSP12 SUPPRESSED LPS-INDUCED LUNG INJURY IN MICE:
To examine whether DUSP12 participates in the pathology of ALI, we first measured changes in the expression of DUSP12 in cells and found that DUSP12 protein and mRNA levels were downregulated in LPS-induced MLECs (Figure 1A, 1B). We established an ALI mouse model via intratracheal LPS administration. We also overexpressed DUSP12 by an adenovirus delivery system 6 h after LPS injection. As shown in Figure 1C and 1D, DUSP12 was upregulated in lung tissue at 24 h after Ad-DUSP12 injection. LPS induced pathological alveolar wall thickness, alveolar collapse and inflammatory cell infiltration, as shown by H&E staining (Figure 1E). DUSP12 overexpression alleviated these pathological changes (Figure 1E). The lung wet weight-to-dry weight ratio (W/D ratio), lung injury score, and proinflammatory cytokine level in alveolar lavage fluid were also ameliorated in the DUSP12-overexpressing group (Figure 1F–1H). These results suggest that increasing the level of DUSP12 can suppress LPS-induced ALI in mice.
DUSP12 KNOCKDOWN AGGRAVATED LPS-INDUCED LUNG INJURY IN MICE:
We then knocked down DUSP12 expression in lung tissue via an adenovirus delivery system 6 h after LPS injection. The expression of DUSP12 was sharply downregulated at 24 h after Ad-shDUSP12 injection (Figure 2A, 2B). LPS-induced pathological alveolar wall thickness, alveolar collapse, and inflammatory cell infiltration were aggravated by DUSP12 knockdown, as shown by H&E staining, an increased lung W/D weight ratio, and an increased lung injury score (Figure 2C–2E). The proinflammatory cytokine level in alveolar lavage fluid was also increased in mice that received Ad-shDUSP12 injection (Figure 2F).
DUSP12 SUPPRESSED LPS-INDUCED PULMONARY ENDOTHELIAL CELL INJURY:
The expression level of DUSP12 was also downregulated in MLECs after LPS insult (Figure 3A). We then overexpressed DUSP12 in pulmonary MLECs (Figure 3B). The transcription levels of proinflammatory cytokines such as TNFα, IL-1, and IL-6 were increased after LPS stimulation. DUSP12 suppressed the transcription of these proinflammatory cytokines (Figure 3C). We also examined the release of these cytokines via ELISA. Consistently, the release of these inflammatory cytokines was inhibited by DUSP12 after LPS stimulation (Figure 3D). Cell apoptosis is a main characteristic of EC injury. We evaluated cell apoptosis via TUNEL staining. After LPS stimulation, the number of TUNEL-positive cells was increased to 19.12% (2.09% in the PBS/Ad-NC group) (Figure 3E). DUSP12 overexpression reduced the percentage of TUNEL-positive cells (10.3%). The same trend was observed regarding caspase-3 activity, which increased after LPS insult and decreased in DUSP12-overexpressing cells (Figure 3F).
DUSP12 SILENCING WORSENED LPS-INDUCED PULMONARY ENDOTHELIAL CELL INJURY:
We examined whether DUSP12 silencing exerted the opposite effects. We knocked down DUSP12 in MLECs with DUSP12 siRNA (Figure 4A). RT-PCR and ELISA were used to evaluate the transcript and protein levels of TNFα, IL-1, and IL-6. LPS elevated the synthesis and release of these proinflammatory factors. Significant upregulation of TNFα, IL-1, and IL-6 was observed in cells transfected with DUSP12 siRNA (Figure 4B, 4C). DUSP12 silencing also increased the percentage of TUNEL-positive cells (17.9% in the scRNA-LPS group; 21.1% in the DUSP12 siRNA group) after LPS insult (Figure 4D). Caspase-3 activity was also increased in the DUSP12 siRNA group compared with the scRNA-LPS group (Figure 4E).
DUSP12 REGULATED OXIDATIVE STRESS:
Excessive oxidative stress causes endothelial cell damage and leads to cell death. We evaluated ROS levels and antioxidant activity in cells after LPS stimulation. Excessive ROS levels, increased NADPH oxidase activity, elevated lipid metabolism intermediate MDA, and decreased SOD2 activity were observed in cells treated with LPS compared with cells in the PBS group. Cells with DUSP12 overexpression exhibited decreased ROS levels, reduced NADPH oxidase activity, decreased MDA levels, and elevated SOD and Gpx activities (Figure 5A, 5B). However, cells with DUSP12 knockdown exhibited higher ROS levels, more NADPH oxidase activity, higher MDA levels, and lower SOD and Gpx activities than those in the scRNA-LPS group (Figure 5C, 5D). These results suggest that DUSP12 protects lung endothelial cells against LPS-induced injury.
DUSP12 AFFECTED ASK1-JNK SIGNALING:
We next investigated the mechanism underlying the protective effects of DUSP12. Studies have reported that DUSP12 can dephosphorylate ASK1, which is upstream of MAPKs. We measured the phosphorylation level of ASK1 in MLECs and found that the phosphorylation of ASK1, as well as the downstream target JNK1/2, was elevated after LPS stimulation. DUSP12 silencing increased the phosphorylation levels of ASK1 and JNK1/2 (Figure 6A, 6B). DUSP12 overexpression reduced the phosphorylation levels of ASK1 and JNK1/2 (Figure 6C, 6D). We then examined whether DUSP12 could directly act on ASK1 in lung endothelial cells. The co-IP results showed that DUSP12 directly bound to ASK1 in lung endothelial cells (Figure 6E).
ASK1-JNK INHIBITION ALLEVIATED THE EFFECTS OF DUSP12 SILENCING:
To confirm that DUSP12 targets ASK1/JNK in LPS-induced endothelial cell injury, we used the JNK1/2 inhibitor SP600125 and ASK1 siRNA to knock down ASK1 (Figure 7A). MLECs were transfected with DUSP12 siRNA and SP600125 or ASK1 siRNA and then treated with LPS. Both the JNK inhibitor and ASK1 siRNA ameliorated DUSP12 knockdown-induced exacerbation, as indicated by downregulated proinflammatory cytokine release, reduced oxidative stress, and decreased percentages of TUNEL-positive cells in the SiDUSP12 +JNK inhibitor and SiDUSP12+ASK1 siRNA groups compared to the Si-DUSP12 group (Figure 7B–7E).
JNK INHIBITION BLOCKED DUSP12 KNOCKDOWN-MEDIATED EXACERBATION IN ALI MICE:
To confirm that JNK is the target of DUSP12 during ALI, JNK activity was inhibited by intraperitoneal injection of SP600125 (15 mg/kg). The JNK inhibitor alone alleviated LPS-induced lung injury and inflammation (Figure 8A–8D). DUSP12 knockdown aggravated pathological alveolar wall thickness, alveolar collapse, and inflammatory cell infiltration, and these effects were all counteracted by the JNK inhibitor, as shown by H&E staining, the lung tissue W/D ratio, injury score, and levels of released TNFα and IL-1 (Figure 8A–8D).
Discussion
Our findings revealed a previously unrecognized role of DUSP12 in protecting against LPS-induced ALI. We observed that DUSP12 levels were decreased in LPS-induced MLECs. DUSP12 overexpression protected against LPS-induced MLEC inflammation and apoptosis, while DUSP12 deficiency exacerbated LPS-induced MLEC inflammation and apoptosis. Moreover, DUSP12 overexpression in vivo also ameliorated LPS-induced ALI in mice.
Cell injury, inflammatory activation, and other complex factors work together to promote the pathogenesis of ALI/ARDS [2]. A variety of cells are involved in this process, including alveolar MLECs, lung endothelial cells, and various inflammatory cells, such as neutrophils and alveolar macrophages [3]. Pulmonary vascular endothelial cells play key roles in lung tissue development, lung tissue immunity, and the maintenance of pulmonary vascular tone and barrier integrity [1]. During the pathology of ALI/ARDS, pulmonary vascular endothelial cells are the first cells exposed to pathological stimuli [13]. Pulmonary vascular endothelial injury causes the occurrence and further progression of ALI/ARDS [1]. A previous study reported that DUSP12 is involved in hepatic lipid metabolism [14] and intracellular bacterial infection [15]. DUSP12 was also shown to alleviate pressure overload-induced cardiac remodeling [4]. In the present study, we found that DUSP12 was involved in the progression of ALI. DUSP12 expression levels changed in LPS-stimulated MLECs. DUSP12 overexpression protected against LPS-induced MLEC injury, while DUSP12 silencing exacerbated MLEC injury. These results suggested that DUSP12 acted against MLEC injury during ALI.
Overproduction of oxidants in the lung tissue is the main cause of ALI [13]. The sources of oxidants may be intrinsic, such as macrophages, neutrophils, and endothelial and alveolar epithelial cells, or the oxidants may be caused by inhaled pollutants or high concentrations of oxygen [16]. In this LPS-induced ALI model, the sources of ROS were mainly activated inflammatory cells and endothelial cells [17]. The complex antioxidant system in the body provides a balanced redox system to maintain the normal function of lung tissue [18]. However, during ALI, increased ROS break this balance and lead to cell injury and apoptosis [19]. In our study, DUSP12 reduced the total cellular ROS level and increased antioxidant activity, such as that of SOD2 and Gpx. This protective effect may be attributed to the protective role of DUSP12 in lung injury.
Previous studies have confirmed the effect of DUSP12 on the ASK1-JNK pathway [20], which is essential in inflammation and oxidative stress in disease [21]. JNK is a mitogen-activated protein kinase (MAPK) family member, and this group also contains extracellular signal-regulated kinase 1/2 (ERK1/2), P38, and ERK5 [21]. MAPKs, which are activated by many factors, such as growth factors, proinflammatory cytokines, and stress, play essential roles in cell development, survival, death, and inflammation [22]. JNK is primarily activated by proinflammatory cytokines and stress by being phosphorylated by MAP3Ks, including ASK1 and TAK1 [22,23]. Studies have shown that the inhibition of MAPKs in ALI mice attenuates inflammation and oxidative stress [24]. Dual-specificity phosphatases are a family of proteins that can dephosphorylate many substrates at threonine/serine residues and/or tyrosine residues [4]. Many of the typical dual-specificity phosphatases contain the MAPK-binding motif and act as MAP kinase phosphatases [25,26]. As an atypical family member, DUSP12 has also been reported to dephosphorylate ASK1 [14]. Our study showed that DUSP12 inhibited ASK1-JNK activation and DUSP12 can directly bind to and dephosphorylate ASK1. This finding was further confirmed with ASK1 siRNA and JNK1/2. Inhibition of the ASK1/JNK pathway counteracted the effect of DUSP12 deficiency. We also confirmed the protective effect of DUSP12 in a mouse ALI model, which provides preclinical data for targeting DUSP12 in ALI/ARDS. Many JNK inhibitors have been proven to be promising in treating ALI [27,28]. However, most of these inhibitors have some off-target effects and may also affect other tissues. In this study, we attempted to use a specific method to target the upstream JNK molecule DUSP12 specifically in lung tissue by an adenovirus delivery system. However, further research is needed before we can develop the most efficiency strategy.
Conclusions
This preliminary study showed that DUSP12 reduced MLEC injury in a murine model of LPS-induced ALI with involvement of the ASK1/JNK pathway. Further studies on the mechanisms of the effects of DUSP12 on epithelial cells are required with investigation of the expression of DUSP12 in human lung tissue samples from cases of ALI/ARDS.
Limitations
Several limitations should be considered. First, it would be desirable to use other ALI model such as cecal ligation, which can better imitate the pathology of ALI caused by secondary infection. Second, MLECs were used instead of human pulmonary microvascular endothelial cells. Although MLECs are widely employed together with the mouse model to explore the pathogenesis and prevention of ALI/ARDS, caution is needed in interpreting results due to inter-species differences.
Figures
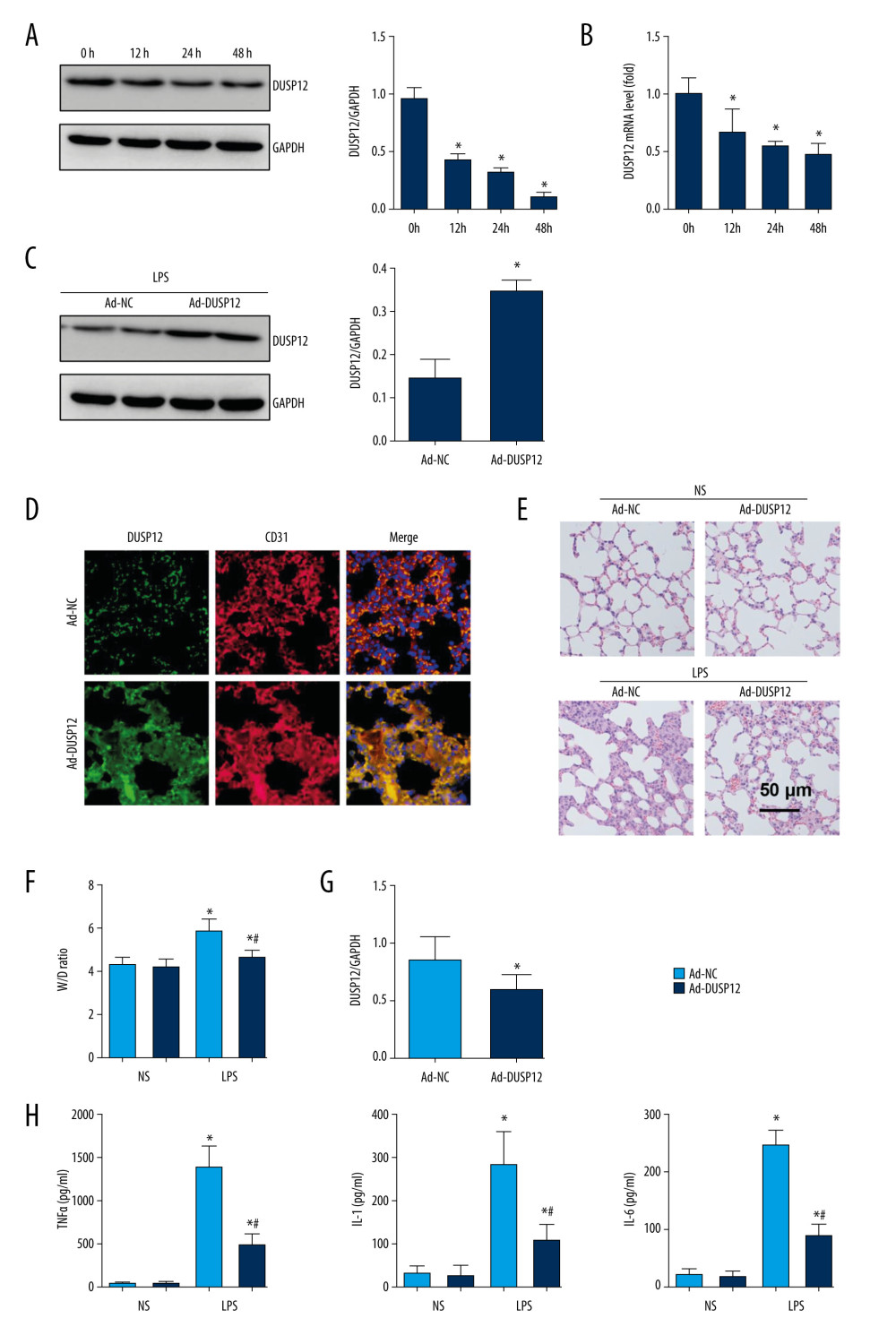 Figure 1. DUSP12 suppressed LPS-induced lung injury in mice. (A, B) DUSP12 protein and mRNA levels in lung tissue after treatment with lipopolysaccharide (LPS) (* P<0.05 vs 0 h). (C) Protein level of DUSP12 in mouse lung tissue 42 h after Ad-DUSP12 injection (n=6). (D) Immunofluorescence staining of CD31 and DUSP12 after Ad-DUSP12 injection (n=5, magnification: 400). (E) Hematoxylin and eosin (H&E) staining (n=5, magnification: 200). (F) Lung wet weight/dry weight ratio (W/D ratio) (n=10). (G) Lung injury score (n=6). (H) Proinflammatory factor levels in alveolar lavage fluid as measured by ELISA (n=6). (* P<0.05 vs Ad-NC/NS; # P<0.05 vs Ad-NC/LPS).
Figure 1. DUSP12 suppressed LPS-induced lung injury in mice. (A, B) DUSP12 protein and mRNA levels in lung tissue after treatment with lipopolysaccharide (LPS) (* P<0.05 vs 0 h). (C) Protein level of DUSP12 in mouse lung tissue 42 h after Ad-DUSP12 injection (n=6). (D) Immunofluorescence staining of CD31 and DUSP12 after Ad-DUSP12 injection (n=5, magnification: 400). (E) Hematoxylin and eosin (H&E) staining (n=5, magnification: 200). (F) Lung wet weight/dry weight ratio (W/D ratio) (n=10). (G) Lung injury score (n=6). (H) Proinflammatory factor levels in alveolar lavage fluid as measured by ELISA (n=6). (* P<0.05 vs Ad-NC/NS; # P<0.05 vs Ad-NC/LPS). 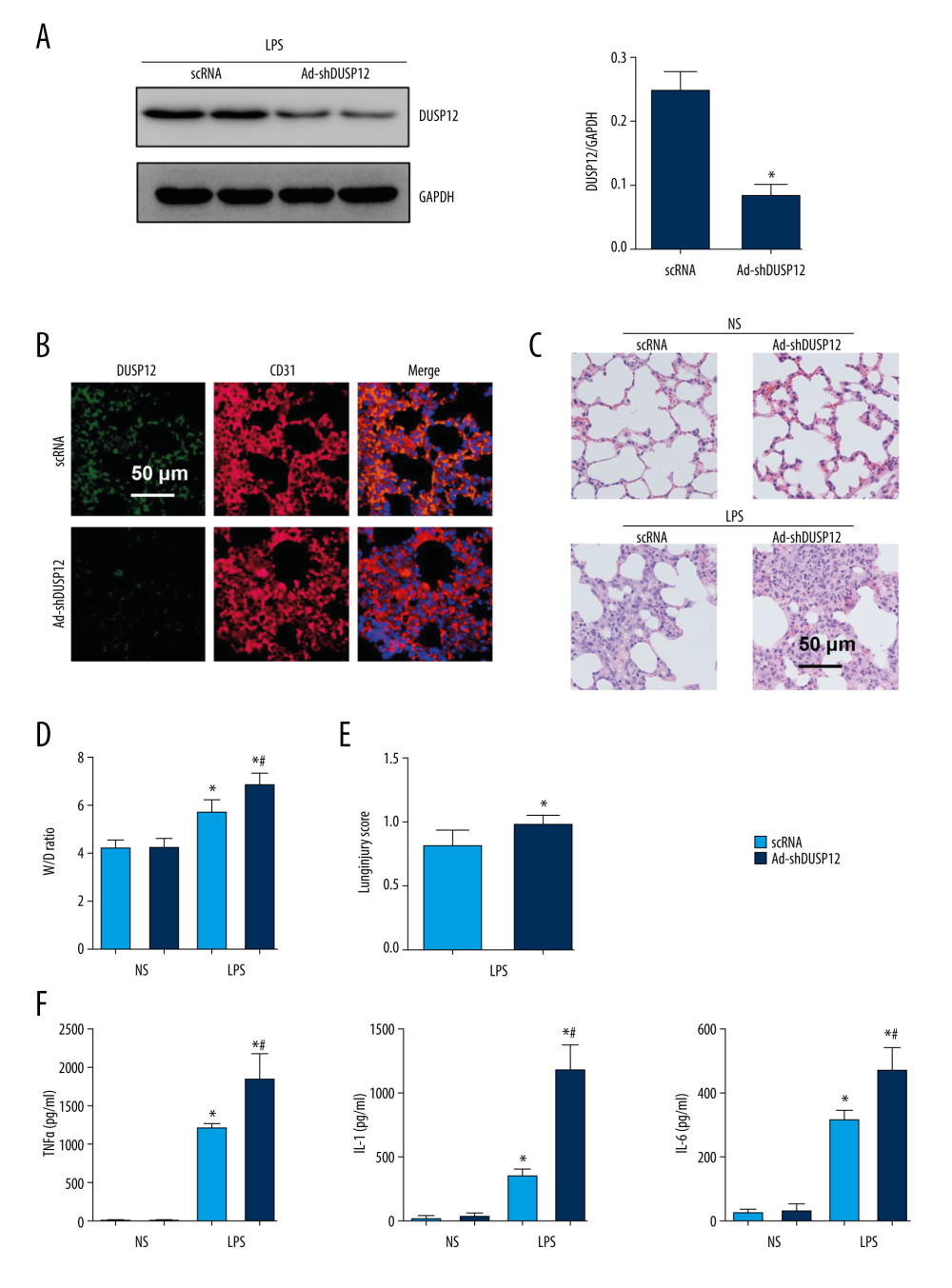 Figure 2. DUSP12 knockdown aggravated LPS-induced lung injury in mice. (A) Protein level of DUSP12 in mouse lung tissue 42 h after Ad-shDUSP12 injection (n=6). (B) Immunofluorescence staining of CD31 and DUSP12 after Ad-shDUSP12 injection (n=5, magnification: 400). (C) H&E staining (n=5, magnification: 200). (D) Lung wet weight/dry weight ratio (W/D ratio) (n=10). (E) Lung injury score (n=6). (F) Proinflammatory factor levels in alveolar lavage fluid as measured by ELISA (n=6) (* P<0.05 vs ScRNA/NS; # P<0.05 vs ScRNA/LPS).
Figure 2. DUSP12 knockdown aggravated LPS-induced lung injury in mice. (A) Protein level of DUSP12 in mouse lung tissue 42 h after Ad-shDUSP12 injection (n=6). (B) Immunofluorescence staining of CD31 and DUSP12 after Ad-shDUSP12 injection (n=5, magnification: 400). (C) H&E staining (n=5, magnification: 200). (D) Lung wet weight/dry weight ratio (W/D ratio) (n=10). (E) Lung injury score (n=6). (F) Proinflammatory factor levels in alveolar lavage fluid as measured by ELISA (n=6) (* P<0.05 vs ScRNA/NS; # P<0.05 vs ScRNA/LPS). 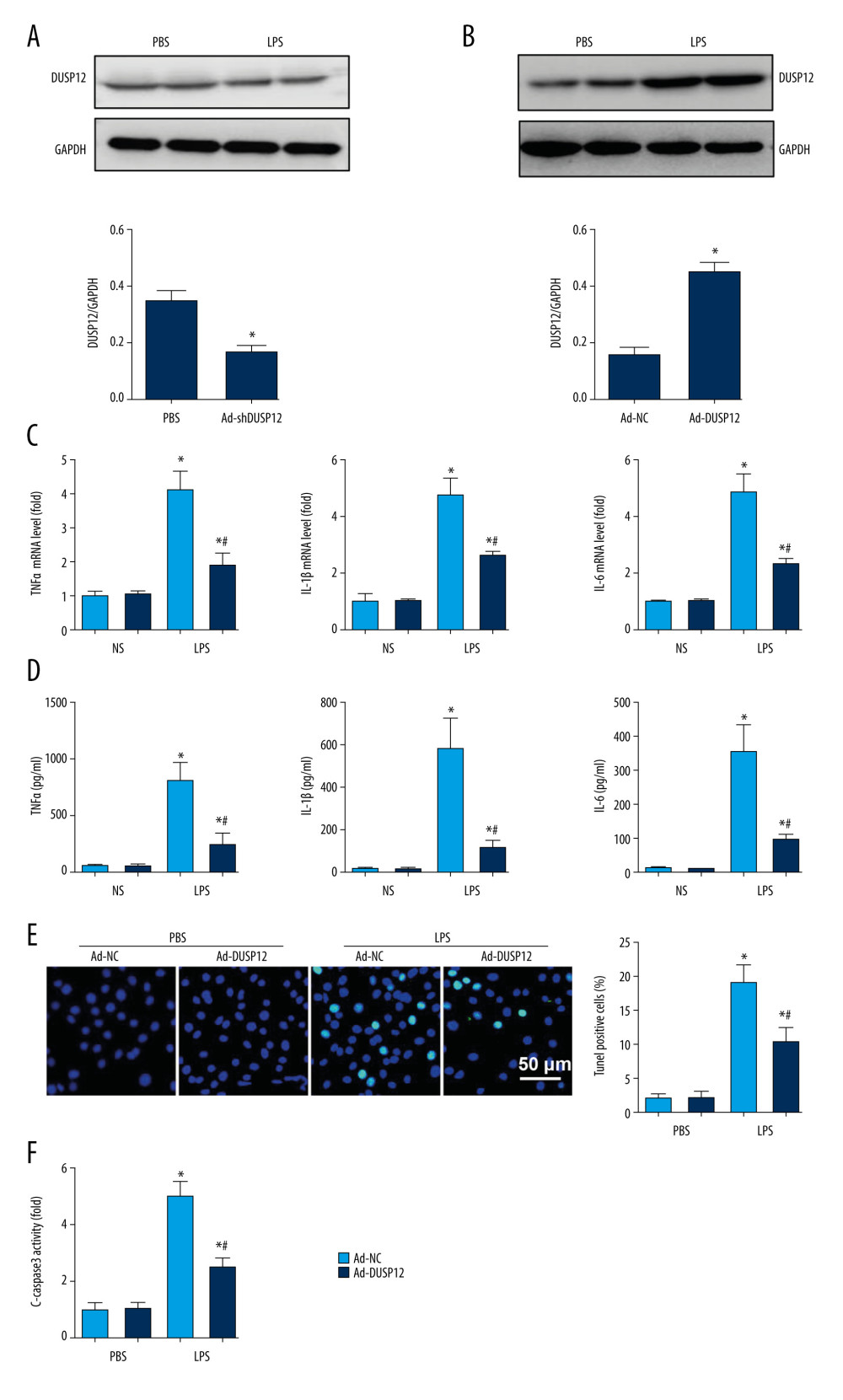 Figure 3. DUSP12 suppressed LPS-induced pulmonary endothelial cell injury. (A) Protein level of DUSP12 in murine lung vascular endothelial cells (MLECs) treated with LPS. (B–F) MLECs were transfected with Ad-DUSP12 and treated with LPS. B. Protein level of DUSP12 in MLECs transfected with Ad-DUSP12. (C) mRNA levels of proinflammatory factors. (D) ELISA measurement of proinflammatory factors. (E) TUNEL staining (magnification: 200). (F) Caspase3 activity (* P<0.05 vs Ad-NC/PBS; # P<0.05 vs Ad-NC/LPS). All in vitro studies were repeated 3 times independently.
Figure 3. DUSP12 suppressed LPS-induced pulmonary endothelial cell injury. (A) Protein level of DUSP12 in murine lung vascular endothelial cells (MLECs) treated with LPS. (B–F) MLECs were transfected with Ad-DUSP12 and treated with LPS. B. Protein level of DUSP12 in MLECs transfected with Ad-DUSP12. (C) mRNA levels of proinflammatory factors. (D) ELISA measurement of proinflammatory factors. (E) TUNEL staining (magnification: 200). (F) Caspase3 activity (* P<0.05 vs Ad-NC/PBS; # P<0.05 vs Ad-NC/LPS). All in vitro studies were repeated 3 times independently. 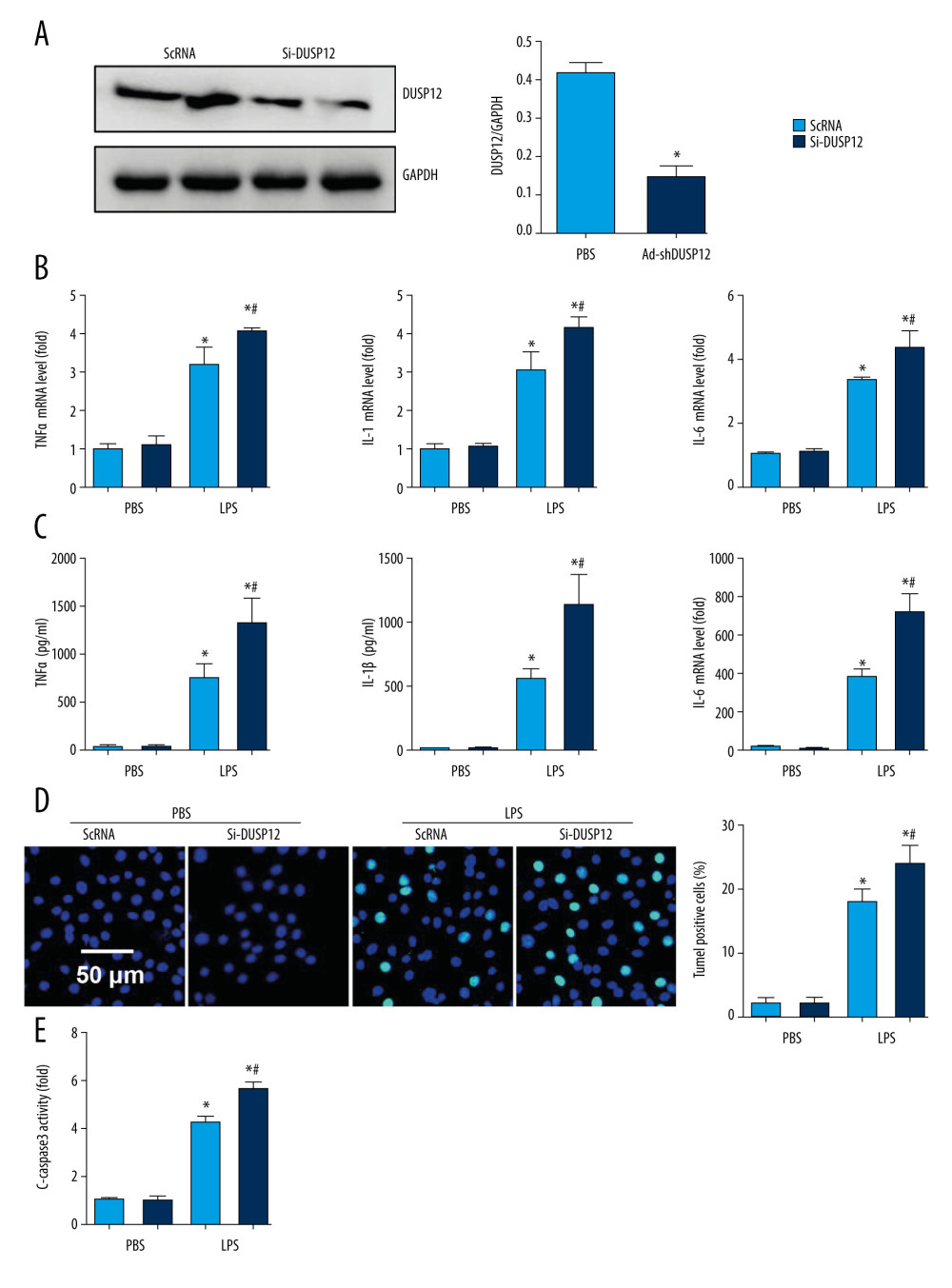 Figure 4. DUSP12 silencing worsened LPS-induced pulmonary endothelial cell injury. (A–E) MLECs were transfected with DUSP12 siRNA and treated with LPS. (A) Protein level of DUSP12 in MLECs transfected with siDUSP12. (B) mRNA levels of proinflammatory factors. (C) ELISA measurement of proinflammatory factors. (D) TUNEL staining (magnification: 200). (E) Caspase-3 activity (* P<0.05 vs ScRNA/PBS; # P<0.05 vs ScRNA/LPS). All in vitro studies were repeated 3 times independently.
Figure 4. DUSP12 silencing worsened LPS-induced pulmonary endothelial cell injury. (A–E) MLECs were transfected with DUSP12 siRNA and treated with LPS. (A) Protein level of DUSP12 in MLECs transfected with siDUSP12. (B) mRNA levels of proinflammatory factors. (C) ELISA measurement of proinflammatory factors. (D) TUNEL staining (magnification: 200). (E) Caspase-3 activity (* P<0.05 vs ScRNA/PBS; # P<0.05 vs ScRNA/LPS). All in vitro studies were repeated 3 times independently. 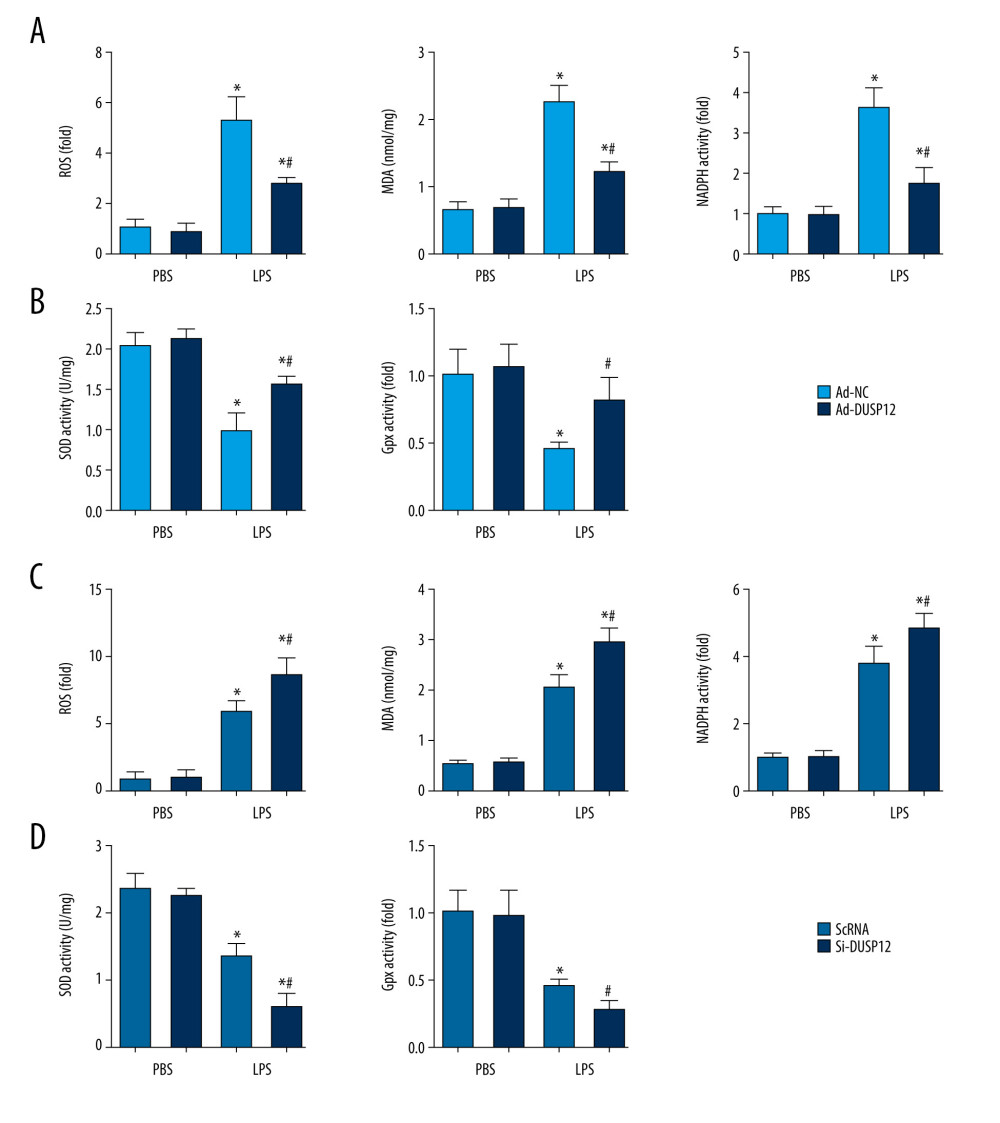 Figure 5. DUSP12 regulated oxidative stress. (A, B) MLECs were transfected with Ad-DUSP12 and treated with LPS. (C, D) MLECs were transfected with DUSP12 siRNA and treated with LPS. (A, C) Reactive oxygen species (ROS) level, malondialdehyde (MDA) level, and nicotinamide adenine dinucleotide phosphate (NADPH) activity in MLECs. (B, D) Superoxide dismutase 2 (SOD2) and glutathione peroxidases (Gpx) activities in MLECs (* P<0.05 vs Ad-NC/PBS or ScRNA/PBS; # P<0.05 vs Ad-NC/LPS or ScRNA/LPS). All in vitro studies were repeated 3 times independently.
Figure 5. DUSP12 regulated oxidative stress. (A, B) MLECs were transfected with Ad-DUSP12 and treated with LPS. (C, D) MLECs were transfected with DUSP12 siRNA and treated with LPS. (A, C) Reactive oxygen species (ROS) level, malondialdehyde (MDA) level, and nicotinamide adenine dinucleotide phosphate (NADPH) activity in MLECs. (B, D) Superoxide dismutase 2 (SOD2) and glutathione peroxidases (Gpx) activities in MLECs (* P<0.05 vs Ad-NC/PBS or ScRNA/PBS; # P<0.05 vs Ad-NC/LPS or ScRNA/LPS). All in vitro studies were repeated 3 times independently. 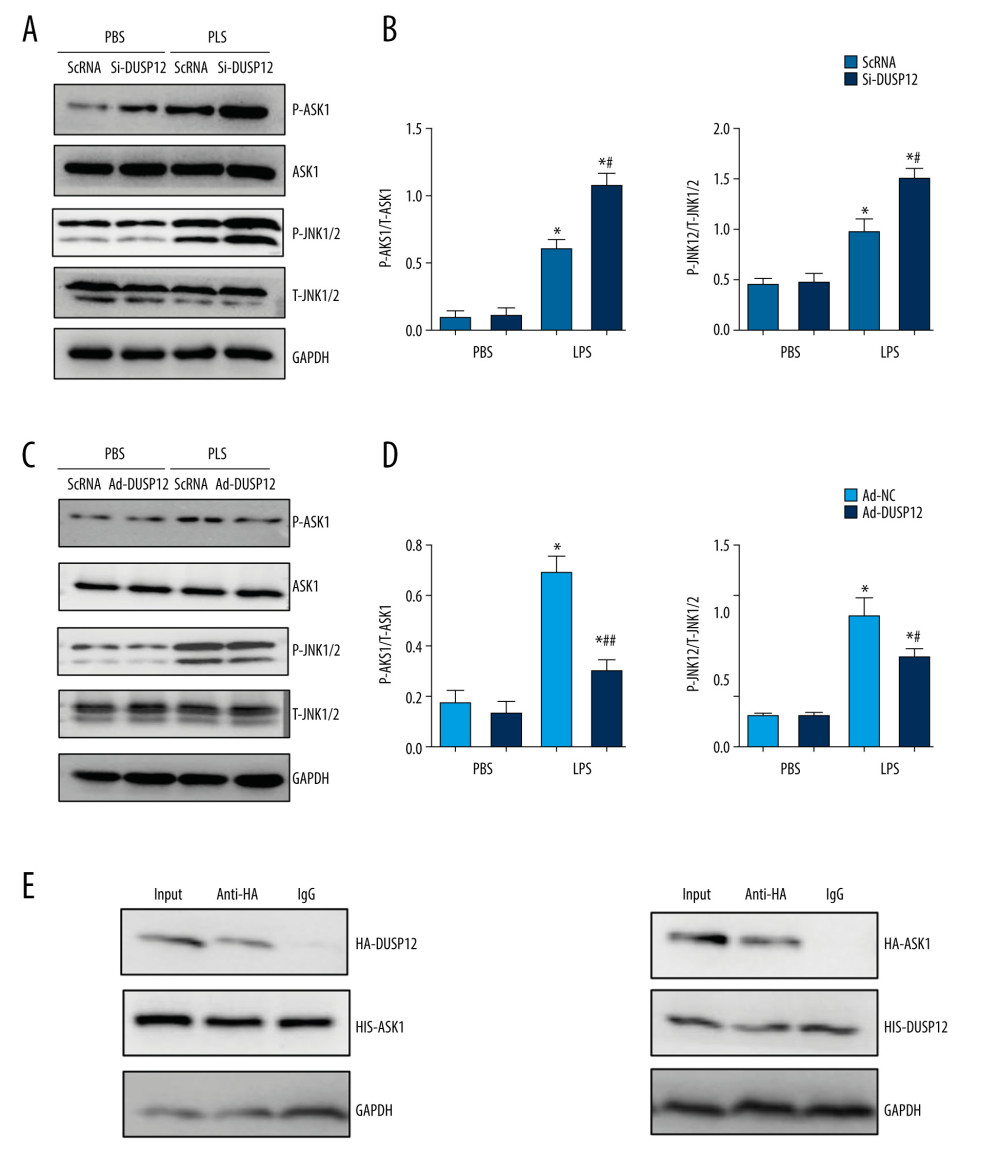 Figure 6. DUSP12 affected ASK1-JNK signaling. (A, B) MLECs were transfected with DUSP12 siRNA and treated with LPS. (A, B) Protein levels of P-ASK1, JNK1/2, T-ASK1, and T-JNK1/2 in MLECs transfected with siDUSP12 (* P<0.05 vs ScRNA/PBS; # P<0.05 vs ScRNA/LPS). (C, D) MLECs were transfected with Ad-DUSP12 and treated with LPS. A and B. Protein levels of P-ASK1, JNK1/2, T-ASK1, and T-JNK1/2 in MLECs transfected with AdDUSP12 (* P<0.05 vs Ad-NC/PBS; # P<0.05 vs Ad-NC/LPS). (E) Co-IP analysis of DUSP12 and ASK1 in MLECs. All in vitro studies were repeated 3 times independently.
Figure 6. DUSP12 affected ASK1-JNK signaling. (A, B) MLECs were transfected with DUSP12 siRNA and treated with LPS. (A, B) Protein levels of P-ASK1, JNK1/2, T-ASK1, and T-JNK1/2 in MLECs transfected with siDUSP12 (* P<0.05 vs ScRNA/PBS; # P<0.05 vs ScRNA/LPS). (C, D) MLECs were transfected with Ad-DUSP12 and treated with LPS. A and B. Protein levels of P-ASK1, JNK1/2, T-ASK1, and T-JNK1/2 in MLECs transfected with AdDUSP12 (* P<0.05 vs Ad-NC/PBS; # P<0.05 vs Ad-NC/LPS). (E) Co-IP analysis of DUSP12 and ASK1 in MLECs. All in vitro studies were repeated 3 times independently. 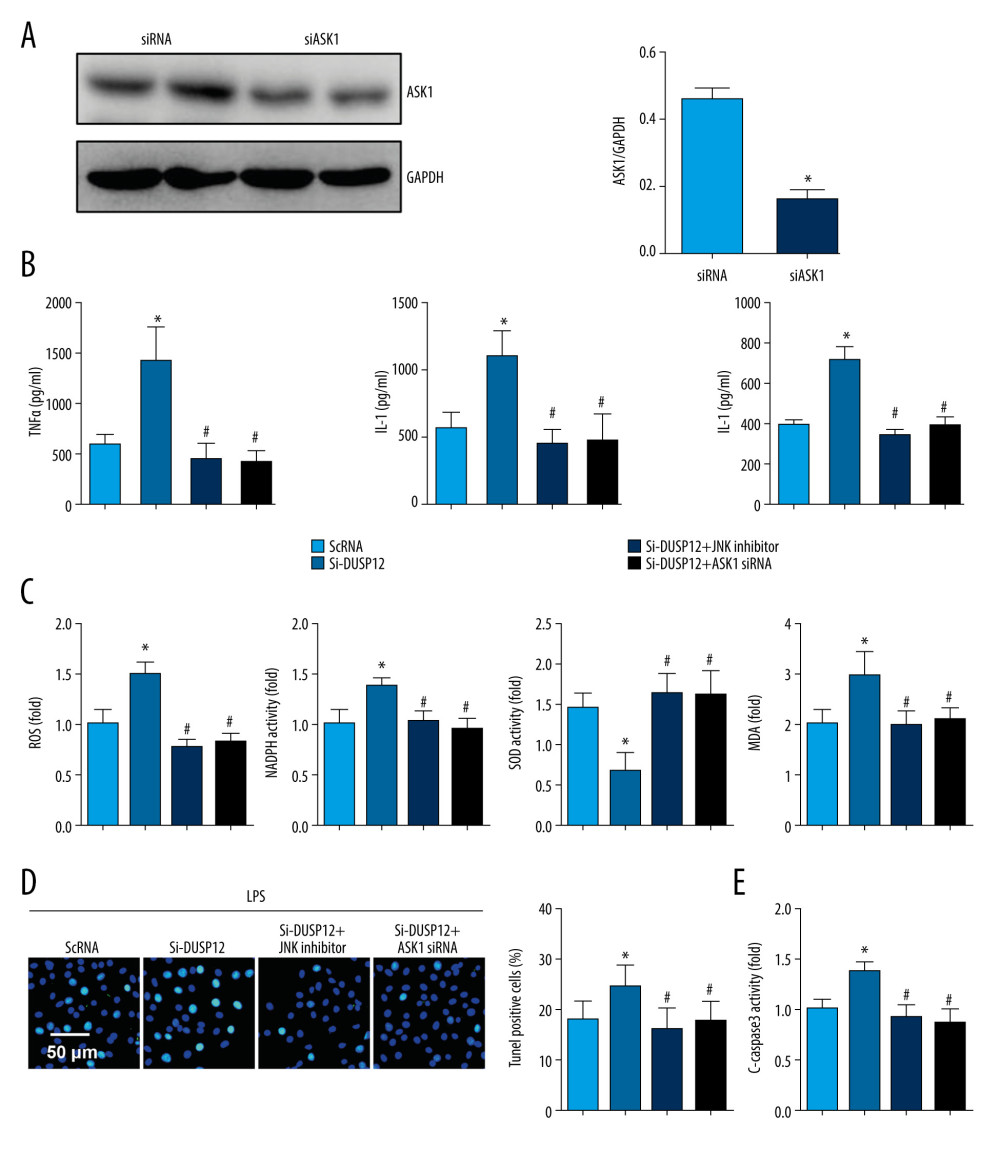 Figure 7. ASK1-JNK inhibition alleviated the effects of DUSP12 silencing. MLECs were transfected with DUSP12 siRNA, treated with the JNK inhibitor or ASK1 siRNA and then treated with LPS. (A) ASK1 protein levels after cells were transfected with ASK1 siRNA. (B) ELISA measurement of proinflammatory factors. (C) ROS level, MDA level, NADPH activity, and SOD activity. (D) TUNEL staining (magnification: 200). (E) Caspase-3 activity (* P<0.05 vs ScRNA/LPS; # P<0.05 vs siDUSP12/LPS). All in vitro studies were repeated 3 times independently.
Figure 7. ASK1-JNK inhibition alleviated the effects of DUSP12 silencing. MLECs were transfected with DUSP12 siRNA, treated with the JNK inhibitor or ASK1 siRNA and then treated with LPS. (A) ASK1 protein levels after cells were transfected with ASK1 siRNA. (B) ELISA measurement of proinflammatory factors. (C) ROS level, MDA level, NADPH activity, and SOD activity. (D) TUNEL staining (magnification: 200). (E) Caspase-3 activity (* P<0.05 vs ScRNA/LPS; # P<0.05 vs siDUSP12/LPS). All in vitro studies were repeated 3 times independently. 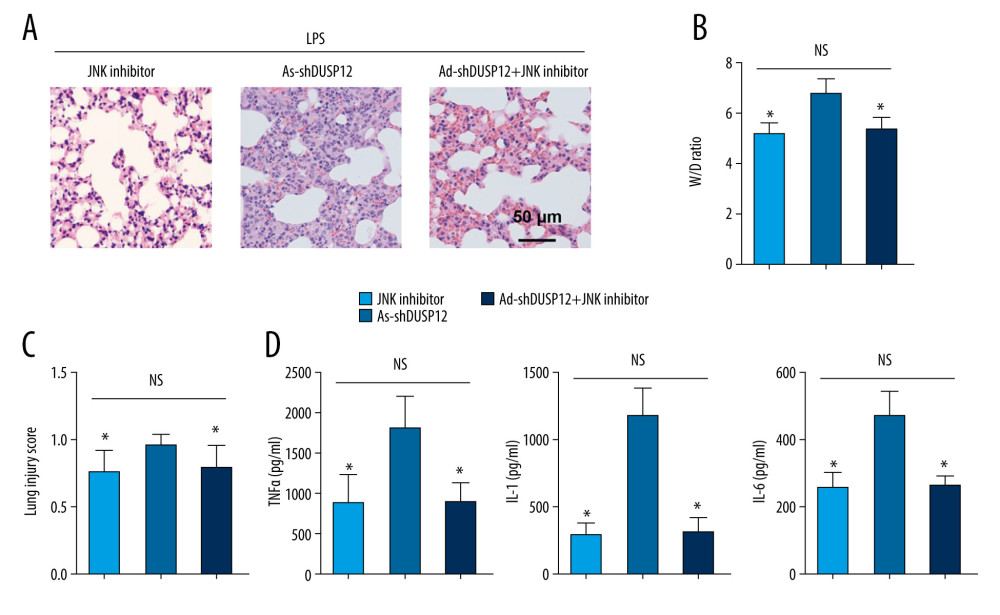 Figure 8. JNK inhibition blocked the DUSP12 knockdown-mediated worsening of ALI in mice. (A) H&E staining (n=5, magnification: 200). (B) Lung wet weight/dry weight ratio (D/W ratio) (n=10). (C) Lung injury score (n=6). (D) Proinflammatory factor levels in alveolar lavage fluid as measured by ELISA (n=6) (* P<0.05 vs Ad-shDUSP12-LPS, NS – not significant).
Figure 8. JNK inhibition blocked the DUSP12 knockdown-mediated worsening of ALI in mice. (A) H&E staining (n=5, magnification: 200). (B) Lung wet weight/dry weight ratio (D/W ratio) (n=10). (C) Lung injury score (n=6). (D) Proinflammatory factor levels in alveolar lavage fluid as measured by ELISA (n=6) (* P<0.05 vs Ad-shDUSP12-LPS, NS – not significant). References
1. Chambers E, Rounds S, Lu Q, Pulmonary endothelial cell apoptosis in emphysema and acute lung injury: Adv Anat Embryol Cell Biol, 2018; 228; 63-86
2. Herrero RSG, Lorente JA, New insights into the mechanisms of pulmonary edema in acute lung injury: Ann Transl Med, 2018; 6; 32
3. Ferruelo APó, Lorente JA, MicroRNAs as biomarkers of acute lung injury: Ann Transl Med, 2018; 6; 34
4. Li WM, Zhao YF, Zhu GF, Dual specific phosphatase 12 ameliorates cardiac hypertrophy in response to pressure overload: Clin Sci (Lond), 2017; 131; 141-54
5. Das SK, Chu WS, Hale TC, Polymorphisms in the glucokinase-associated, dual-specificity phosphatase 12 (DUSP12) gene under chromosome 1q21 linkage peak are associated with type 2 diabetes: Diabetes, 2006; 55; 2631-39
6. Monteiro LF, Forti FL, Network analysis of DUSP12 partners in the nucleus under genotoxic stress: J Proteomics, 2019; 197; 42-52
7. Picozza M, Avitabile D, Magenta A, Monocyte dysfunction induced by low density lipoprotein occurs via a DUSP-1/p38 MAPK signaling impairment: Int J Cardiol, 2018; 255; 166-67
8. Kozarova A, Hudson JW, Vacratsis PO, The dual-specificity phosphatase hYVH1 (DUSP12) is a novel modulator of cellular DNA content: Cell Cycle, 2011; 10; 1669-78
9. Hu CZR, Wang C, Ma X, Lack of association between genetic polymorphisms within DUSP12 – ATF6 locus and glucose metabolism related traits in a Chinese population: BMC Med Genet, 2011; 12; 3
10. Geng Q, Xhabija B, Knuckle C, The atypical dual specificity phosphatase hYVH1 associates with multiple ribonucleoprotein particles: J Biol Chem, 2017; 292; 539-50
11. Matute-Bello G, Downey G, Moore BB, An official American Thoracic Society workshop report: Features and measurements of experimental acute lung injury in animals: Am J Respir Cell Mol Biol, 2011; 44; 725-38
12. Lim YC, Garcia-Cardena G, Allport JR, Heterogeneity of endothelial cells from different organ sites in T-cell subset recruitment: Am J Pathol, 2003; 162; 1591-601
13. Gonzalez H, Horie S, Laffey JG, Emerging cellular and pharmacologic therapies for acute respiratory distress syndrome: Curr Opin Crit Care, 2021; 27; 20-28
14. Huang Z, Wu LM, Zhang JL, Dual specificity phosphatase 12 regulates hepatic lipid metabolism through inhibition of the lipogenesis and apoptosis signal-regulating kinase 1 pathways: Hepatology, 2019; 70; 1099-118
15. Cho SSL, Han J, James SJ, Dual-specificity phosphatase 12 targets p38 MAP kinase to regulate macrophage response to intracellular bacterial infection: Front Immunol, 2017; 8; 1259
16. Kellner M, Noonepalle S, Lu Q, ROS signaling in the pathogenesis of acute lung injury (ALI) and acute respiratory distress syndrome (ARDS): Adv Exp Med Biol, 2017; 967; 105-37
17. Kurundkar D, Kurundkar AR, Bone NB, SIRT3 diminishes inflammation and mitigates endotoxin-induced acute lung injury: JCI Insight, 2019; 4; e120722
18. Ornatowski WLQ, Yegambaram M, Garcia AE, Complex interplay between autophagy and oxidative stress in the development of pulmonary disease: Redox Biol, 2020; 36; 101679
19. Cross CE, Eiserich JP, Oxidative stress in acute lung injury: Deja vu or something new?: Crit Care Med, 2004; 32; 892-93
20. Qiu T, Wang T, Zhou J, DUSP12 protects against hepatic ischemia-reperfusion injury dependent on ASK1-JNK/p38 pathway in vitro and in vivo: Clin Sci (Lond), 2020; 134; 2279-94
21. Li G, Qi W, Li X, Recent advances in c-Jun N-terminal kinase (JNK) inhibitors: Curr Med Chem, 2021; 28; 607-27
22. Kim EK, Choi EJ, Pathological roles of MAPK signaling pathways in human diseases: Biochim Biophys Acta, 2010; 1802; 396-405
23. Ha J, Kang E, Seo J, Phosphorylation dynamics of JNK signaling: Effects of dual-specificity phosphatases (DUSPs) on the JNK pathway: Int J Mol Sci, 2019; 20; 6157
24. Huang WCLC, Liang YT, Hung HC, Phloretin attenuates LPS-induced acute lung injury in mice via modulation of the NF-κB and MAPK pathways: Int Immunopharmacol, 2016; 40; 98-105
25. Chuang HC, Tan TH, MAP4K Family kinases and DUSP family phosphatases in T-cell signaling and systemic lupus erythematosus: Cells, 2019; 8; 1433
26. Yin R, Eger G, Sarri N, Dual specificity phosphatase (DUSP)-4 is induced by platelet-derived growth factor- BB in an Erk1/2-, STAT3- and p53-dependent manner: Biochem Biophys Res Commun, 2019; 519; 469-74
27. Shen H, Wu N, Wang Y, JNK inhibitor SP600125 attenuates paraquat-induced acute lung injury: An in vivo and in vitro study: Inflammation, 2017; 40; 1319-30
28. Lou L, Hu D, Chen S, Protective role of JNK inhibitor SP600125 in sepsis-induced acute lung injury: Int J Clin Exp Pathol, 2019; 12; 528-38w
Figures
Figure 1. DUSP12 suppressed LPS-induced lung injury in mice. (A, B) DUSP12 protein and mRNA levels in lung tissue after treatment with lipopolysaccharide (LPS) (* P<0.05 vs 0 h). (C) Protein level of DUSP12 in mouse lung tissue 42 h after Ad-DUSP12 injection (n=6). (D) Immunofluorescence staining of CD31 and DUSP12 after Ad-DUSP12 injection (n=5, magnification: 400). (E) Hematoxylin and eosin (H&E) staining (n=5, magnification: 200). (F) Lung wet weight/dry weight ratio (W/D ratio) (n=10). (G) Lung injury score (n=6). (H) Proinflammatory factor levels in alveolar lavage fluid as measured by ELISA (n=6). (* P<0.05 vs Ad-NC/NS; # P<0.05 vs Ad-NC/LPS).Figure 2. DUSP12 knockdown aggravated LPS-induced lung injury in mice. (A) Protein level of DUSP12 in mouse lung tissue 42 h after Ad-shDUSP12 injection (n=6). (B) Immunofluorescence staining of CD31 and DUSP12 after Ad-shDUSP12 injection (n=5, magnification: 400). (C) H&E staining (n=5, magnification: 200). (D) Lung wet weight/dry weight ratio (W/D ratio) (n=10). (E) Lung injury score (n=6). (F) Proinflammatory factor levels in alveolar lavage fluid as measured by ELISA (n=6) (* P<0.05 vs ScRNA/NS; # P<0.05 vs ScRNA/LPS).Figure 3. DUSP12 suppressed LPS-induced pulmonary endothelial cell injury. (A) Protein level of DUSP12 in murine lung vascular endothelial cells (MLECs) treated with LPS. (B–F) MLECs were transfected with Ad-DUSP12 and treated with LPS. B. Protein level of DUSP12 in MLECs transfected with Ad-DUSP12. (C) mRNA levels of proinflammatory factors. (D) ELISA measurement of proinflammatory factors. (E) TUNEL staining (magnification: 200). (F) Caspase3 activity (* P<0.05 vs Ad-NC/PBS; # P<0.05 vs Ad-NC/LPS). All in vitro studies were repeated 3 times independently.Figure 4. DUSP12 silencing worsened LPS-induced pulmonary endothelial cell injury. (A–E) MLECs were transfected with DUSP12 siRNA and treated with LPS. (A) Protein level of DUSP12 in MLECs transfected with siDUSP12. (B) mRNA levels of proinflammatory factors. (C) ELISA measurement of proinflammatory factors. (D) TUNEL staining (magnification: 200). (E) Caspase-3 activity (* P<0.05 vs ScRNA/PBS; # P<0.05 vs ScRNA/LPS). All in vitro studies were repeated 3 times independently.Figure 5. DUSP12 regulated oxidative stress. (A, B) MLECs were transfected with Ad-DUSP12 and treated with LPS. (C, D) MLECs were transfected with DUSP12 siRNA and treated with LPS. (A, C) Reactive oxygen species (ROS) level, malondialdehyde (MDA) level, and nicotinamide adenine dinucleotide phosphate (NADPH) activity in MLECs. (B, D) Superoxide dismutase 2 (SOD2) and glutathione peroxidases (Gpx) activities in MLECs (* P<0.05 vs Ad-NC/PBS or ScRNA/PBS; # P<0.05 vs Ad-NC/LPS or ScRNA/LPS). All in vitro studies were repeated 3 times independently.Figure 6. DUSP12 affected ASK1-JNK signaling. (A, B) MLECs were transfected with DUSP12 siRNA and treated with LPS. (A, B) Protein levels of P-ASK1, JNK1/2, T-ASK1, and T-JNK1/2 in MLECs transfected with siDUSP12 (* P<0.05 vs ScRNA/PBS; # P<0.05 vs ScRNA/LPS). (C, D) MLECs were transfected with Ad-DUSP12 and treated with LPS. A and B. Protein levels of P-ASK1, JNK1/2, T-ASK1, and T-JNK1/2 in MLECs transfected with AdDUSP12 (* P<0.05 vs Ad-NC/PBS; # P<0.05 vs Ad-NC/LPS). (E) Co-IP analysis of DUSP12 and ASK1 in MLECs. All in vitro studies were repeated 3 times independently.Figure 7. ASK1-JNK inhibition alleviated the effects of DUSP12 silencing. MLECs were transfected with DUSP12 siRNA, treated with the JNK inhibitor or ASK1 siRNA and then treated with LPS. (A) ASK1 protein levels after cells were transfected with ASK1 siRNA. (B) ELISA measurement of proinflammatory factors. (C) ROS level, MDA level, NADPH activity, and SOD activity. (D) TUNEL staining (magnification: 200). (E) Caspase-3 activity (* P<0.05 vs ScRNA/LPS; # P<0.05 vs siDUSP12/LPS). All in vitro studies were repeated 3 times independently.Figure 8. JNK inhibition blocked the DUSP12 knockdown-mediated worsening of ALI in mice. (A) H&E staining (n=5, magnification: 200). (B) Lung wet weight/dry weight ratio (D/W ratio) (n=10). (C) Lung injury score (n=6). (D) Proinflammatory factor levels in alveolar lavage fluid as measured by ELISA (n=6) (* P<0.05 vs Ad-shDUSP12-LPS, NS – not significant). In Press
Clinical Research
Effects of Single-Bout Endurance Exercise Intensity on Peripheral Neurotrophic Factors in Patients With Isc...Med Sci Monit In Press; DOI: 10.12659/MSM.952089
Review article
Anisodus tanguticus in Cancer Research: A Review of Traditional Use, Phytochemistry, Extraction Methods, an...Med Sci Monit In Press; DOI: 10.12659/MSM.952999
Clinical Research
Nasal Mucociliary Clearance and Its Relationship With Disease Severity in Patients With Multiple SclerosisMed Sci Monit In Press; DOI: 10.12659/MSM.952850
Clinical Research
Modified Thoracoabdominal Nerves Block Through the Perichondrial Approach vs Subcostal Transversus Abdomini...Med Sci Monit In Press; DOI: 10.12659/MSM.953976
Most Viewed Current Articles
17 Jan 2024 : Review article 14,176,570
Vaccination Guidelines for Pregnant Women: Addressing COVID-19 and the Omicron VariantDOI :10.12659/MSM.942799
Med Sci Monit 2024; 30:e942799
13 Nov 2021 : Clinical Research 3,762,188
Acceptance of COVID-19 Vaccination and Its Associated Factors Among Cancer Patients Attending the Oncology ...DOI :10.12659/MSM.932788
Med Sci Monit 2021; 27:e932788
14 Dec 2022 : Clinical Research 2,466,310
Prevalence and Variability of Allergen-Specific Immunoglobulin E in Patients with Elevated Tryptase LevelsDOI :10.12659/MSM.937990
Med Sci Monit 2022; 28:e937990
16 May 2023 : Clinical Research 708,927
Electrophysiological Testing for an Auditory Processing Disorder and Reading Performance in 54 School Stude...DOI :10.12659/MSM.940387
Med Sci Monit 2023; 29:e940387