16 April 2022: Database Analysis
Identification of Novel Genes and Associated Drugs in Cervical Cancer by Bioinformatics Methods
Dan Wang1CDE, Yanling Liu2F, Shuyu Cheng1D, Guoyan Liu123AG*DOI: 10.12659/MSM.934799
Med Sci Monit 2022; 28:e934799






Abstract
BACKGROUND: Cervical cancer is one of the common gynecological tumors that seriously harm women’s health, so it is particularly important to accurately explore the underlying mechanism of its occurrence and clinical prognosis.
MATERIAL AND METHODS: In the GEO database, GEO2R was used to analyze the differentially expressed genes from the 4 databases: GSE6791, GSE9750, GSE63514, and GSE67522. Then, the DAVID website was used to perform Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses. These protein–protein interaction (PPI) networks of DEGS were visualized and analyzed using the STRING website and the hub genes were further screened using the Cytohubba plugin. Lastly, the functions of the hub genes were further analyzed by Gene Expression Profiling Interactive Analysis (GEPIA) online tools, Human Protein Atlas (HPA) databases, and the QuartataWeb database.
RESULTS: In the 4 Profile datasets, 101 cancer tissues and 67 normal tissues were collected. Among the 78 differentially expressed genes in the 4 datasets, 51 genes were upregulated and 27 genes were downregulated. The PPIs of these differentially expressed genes were visualized using Cytoscape and the Interaction Gene Search Tool (STRING). Then, further analysis of hub genes using the GEPIA tool and Kaplan-Meier curves that showed upregulation of CDK1 and PRC1 is associated with better survival, while AURKA is associated with worse survival. Among these hub genes, only AURKA was closely related to the prognosis of cervical cancer, and 21 potential drugs were found.
CONCLUSIONS: These results suggest that AURKA and its drug candidates can improve the individualized diagnosis and treatment of cervical cancer in the future.
Keywords: Biomarkers, Pharmacological, Biomarkers, Tumor, Aurora Kinase A, Computational Biology, Databases, Genetic, Female, Gene Regulatory Networks, Humans, Protein Interaction Maps
Background
Cervical cancer is the most common malignant tumor of the female reproductive system. In recent years, cervical cytological screening, HPV testing, colposcopy, and histopathology have become widely available to facilitate early detection and treatment of cervical cancer [1]. However, the cure rate for cervical cancer remains low in many developing countries [2]. The majority of cervical cancers are linked to the human papillomavirus, which is a spherical DNA virus that causes the squamous epithelium of the skin’s mucous membranes to proliferate [3]. Despite the efforts of the human papillomavirus vaccination program, the incidence of cervical cancer remains very high [4]. Currently, cervical cancer screening, surgical techniques, radiotherapy equipment, chemotherapy, and other treatment methods have been relatively effective, but the clinical treatment effect for advanced and recurrent cervical cancer is still far from sufficient, resulting in poor prognosis of patients. With the development of modern molecular biology and genomics, targeted therapy has become one of the research hotspots in the treatment of advanced or recurrent cervical cancer, and has achieved good clinical efficacy in the treatment of cervical cancer. Current targeted therapies for cervical cancer mainly include antiangiogenic drugs, poly-ADP-ribose polymerase (PARP) inhibitors, immune checkpoint inhibitors, and epithelial growthfactor (EGF) receptor blockers [5]. Although these new prevention methods have made great strides in combination with traditional treatments, the 5-year disease-free survival rate for patients with advanced cervical cancer is only 45% [6]. A great deal of attention has been paid to the pathophysiology of cervical cancer, but the exact pathogenesis of cervical cancer is still not clear. Hence, an effective method is badly needed to explore the internal mechanism and targeted drugs affecting the occurrence of cervical cancer.
High-throughput sequencing has been widely used to study the pathological mechanism of diseases, especially in the prevention of cancer [7–9]. A variety of public databases are available for clinical and sequencing data download and analysis [10]. Reanalysis of the gene expression profiles associated with cervical cancer is important for understanding the pathogenesis and molecular mechanisms of cervical cancer [11]. Various studies have explored the pathogenic genes involved in cervical cancer. Dan et al identified PLOD2, ANLN, AURKA, and AR as possible therapeutic targets of cervical cancer through comprehensive analysis of cervical cancer-related gene expression profiles combined with bioinformatics [12]. Dan et al found that CDC45, GINS2, MCM2, and PCNA may be related to the occurrence and development of cervical cancer, and further verified that MCM2 knockdown can inhibit the proliferation of cervical cancer cells [13]. Although many studies have focused on certain genes in cervical cancer, it is still far from enough to reveal the underlying pathogenesis [14–16].Therefore, the present study used bioinformatics methods to find novel prognostic biomarkers of cervical cancer.
The DEGs in expression profiles GSE6791, GSE9750, GSE63514, and GSE67522 from the Gene Expression Omnibus were identified first in the present work. Then, the DEGs between cervical cancer and healthy groups were analyzed. GO annotation, KEGG pathway annotation, and PPI analysis were conducted among the DEGs using several bioinformatics methods. Further analysis of the clinical significance of these hub genes (including expression and survival analysis) via the Gene Expression Omnibus may provide new directions for understanding the role of these hub genes in the development and metastasis of cervical cancer. Discovery and development of new cancer drugs is expensive and time consuming. Our study aims to explore the targeted therapy of cervical cancer genes and related drugs, which will provide new ideas for drug use in the treatment of cervical cancer through the new use of old drugs.
Material and Methods
MICROARRAY DATA INFORMATION:
By searching for cervical cancer in the Gene Expression Omnibus (GEO;
DATA IDENTIFICATION OF DEGS:
DEGs between cervical cancer specimens and normal specimens were identified via GEO2R online tools (
FUNCTIONAL ENRICHMENT ANALYSIS:
Gene ontology analysis (GO) is a widely used method to identify biological properties of genes. It mainly includes the biological processes that genes participate in, the cell locations they are found in, and the molecular functions they play [17]. The Kyoto Encyclopedia of Genes and Genomes (KEGG) serves as a collection for assessing the signaling pathways of disease-related genes [18]. DAVID (https://david.ncifcrf.gov/) is a bioinformatics database that is used to analyze differential gene function and pathway enrichment analysis [19]. Therefore, the DAVID website was used for GO and KEGG analysis of differentially expressed genes (P<0.05).
PPI NETWORK OF DEGS:
The Retrieval of Interacting Genes (STRING: http://string-db.org) was used to determine the interaction among the DEGs [20]. The results with a minimum interaction score of 0.9 were analyzed and visualized in Cytoscape software (version 3.6.1; https://cytoscape.org). All parameters in cytoHubba were set by default. Nodes with disconnected PPI network are hidden. The top 10 hub genes from the PPI network were selected using the plugin cytoHubba [21] (score >15).
EXPRESSION ANALYSIS OF HUB GENES IN CERVICAL CANCER:
The expression of these hub genes in cervical tissue was examined using GEPIA2 (http://gepia2.cancerpku.cn/), a web-based tool that interactively analyzes gene expression profiles in cancer patients and normal patients from TCGA and GTEX data [22]. Kaplan-Meier curves were used to analyze the correlation between hub genes and overall survival in patients with cervical cancer [23]. The protein expression profile of these hub genes was obtained through the HPA database (https://www.proteinatlas.org/), which is a transcriptome- and proteomics-based technique that studies protein expression in different human tissues and organs at RNA and protein levels [24]. We further analyzed the expression of hub gene in cervical cancer tissues and normal tissues by using immunohistochemical data from the HPA database.
FUNCTION AND PROGNOSIS ANALYSIS OF HUB GENES IN CERVICAL CANCER:
cBioPortal (http://www.cbioportal.org) is a visual tool for studying and analyzing cancer genetic data [25]. The mutation rate and mutation form of these hub genes were analyzed using the cBioportal tool. The GEPIA2 database was again used to analyze the relationship between these hub genes and the clinicopathological staging of cervical cancer.
THE CONSTRUCTION OF DRUG-GENE INTERACTION:
QuartataWeb (http://quartata.csb.pitt.edu/) [26] is a comprehensive online platform for composite genomics, which can be used to search for and infer drug-target interactions, target cell association pathways, and drug-cell pathway associations. The top 10 hub genes were uploaded into the database to screen existing drugs or compounds.
STATISTICS ANALYSIS:
All analyses were performed using GraphPad Prism version 6.0 (GraphPad Software, USA). Data are presented as mean±standard deviation, and differences between groups were analyzed using the
Results
IDENTIFICATION OF DEGS:
DEGs (2103 in GSE6791, 815 in GSE9750, 1977 in GSE63514, and 427 in GSE67522) were identified by GEO2R (Figure 1A–1D). The overlap in the 4 datasets contained 78 genes, as showed in the Venn diagram (Figure 1E, 1F). As a result, 51 upregulated genes and 27 downregulated genes were obtained (Table 1).
FUNCTIONAL ENRICHMENT ANALYSIS OF DEGS:
The 78 overlapping DEGs were subjected to the GO enrichment and KEGG pathway analysis. In GO terms of Biological Process (BP) annotation (Figure 2A), it was mainly enriched in the “DNA replication initiation”, “cell division”, “cell proliferation”, “DNA replication”, “G1/S transition of mitotic cell cycle”, “mitotic nuclear divsion”, “mitotic cytokinesis”, and “G2/M transition of mitotic cell cycle”. In Cellular Component (CC) annotation (Figure 2B), it was enriched in the “nucleus”, “nucleoplasm”, “cytosol”, “midbody”, “centrosome”, “spindle”, “microtubule”, “spindle pole”, “nuclear chromosome, telomeric region”, “spindle microtubule”, “mitotic spindle”, and “MCM complex”. In Molecular Function (MF) annotation (Figure 2C), it was enriched in the “protein binding”, “single-stranded DNA binding”, “ATP binding”, “DNA helicase activity”, and “protein kinase binding”. Additionally, KEGG analysis showed that these DEGs were mainly enriched in the “cell cycle” and “DNA replication” (Figure 2D).
THE CONSTRUCTION OF PPI AND HUB GENES ANALYSIS:
To better explore the role of these DEGs in cervical cancer, we analyzed 78 DEGs using the STRING database (Figure 3A). There were 41 nodes and 218 edges enriched in the PPI network. The top 10 hub genes were selected using cytoHubba. As shown in Figure 3B, the top 10 hub genes were: CDK1, CDC20, AURKA, TOP2A, ASPM, NCAPG, KIF23, CENPF, KIF20A, and PRC1. All parameters in cytoHubba were set by default.
CLINICAL SIGNFICANCE OF 10 HUB GENES:
To further identify the reliability of the hub genes in cervical cancer, we searched the expression levels and prognostic information about these hub genes for cervical cancer via GEPIA2. All 10 hub genes (CDK1, CDC20, AURKA, TOP2A, ASPM, NCAPG, KIF23, CENPF, KIF20A, and PRC1) were reconfirmed via GEPIA2. Compared with normal cervical tissues, the expression level of these DEGs all was higher (Figure 4). Moreover, overall survival rate with higher CDK1 and PRC1 was significantly higher. However, lower AURKA was associated with higher overall survival rate (Figure 5A–5C), suggesting that CDK1, AURKA, and PRC1 play pivotal roles in the process of cervical cancer. Furthermore, the protein expressions of CDK1, AURKA, and PRC1 were significantly upregulated in cervical cancer compared to normal tissues. Among these highly expressed genes, CDK1 and AURKA spread to the cytoplasm in cervical cancer tissues, while PRC1 distribution showed no significant difference (Figure 6).
The occurrence of cancer depends on the accumulation of gene mutations. Due to the rapid cell division and abnormal DNA damage repair mechanism when cancer occurs, the genome of cancer cells becomes more unstable and prone to gene mutations. Therefore, we examined the mutation rate of these hub genes and analyzed their role in cervical cancer via the cBioPortal database. Our analysis of the mutation rate of the hub genes found that ASPM and CENPF had the highest genetic mutation rates, and CDK1, AURKA, and PRC1 were 0.7%, 2.9%, and 2.2%, respectively (Figure 7A). Among the hub gene mutations, CDK1 mutations were mainly shallow deletions, the AURKA and PRC1 mutations were mainly gene amplification (Figure 7B). Clinicopathological stage is an important index to evaluate the range of invasion and metastasis, as well as the degree of disease progression, metastasis, and prognosis of malignant tumors. Here, we examined 3 genes (CDK, AURKA, and PRC1) that were significantly associated with clinical prognosis. Notably, only AURKA expression increased with the expansion of cervical cancer invasion and metastasis (Figure 7C).
THE CONSTRUCTION OF DRUG-GENE INTERACTION:
Three genes associated with survival were uploaded into QuartataWeb for drug-gene interaction analysis. As shown in Table 2, AURKA was identified and matched with 21 predicted drugs that are FDA-approved: Fostamatinib, Regorafenib, Dasatinib, Brigatinib, Ponatinib, Pazopanib, Sorafenib, Imatinib, Nintedanib, Acetylsalicylic acid, Sunitinib, Bosutinib, Tamoxifen, Dabrafenib, Vandetanib, Lenvatinib, Cabozantinib, Baricitinib, Axitinib, Tofacitinib, and Midostaurin are shared by these genes. Thus, we speculate that these drugs could play a role in the treatment of cervical cancer.
Discussion
Cervical cancer is a malignant tumor originating from the cervix and is the most widespread malignant tumor of the female reproductive system. At present, cervical cancer ranks fourth among female malignant tumors in the world [27]. The development of cervical cancer is a complex process related to a variety of genetic and environmental factors. Although the development of the HPV vaccine has improved prevention and treatment of cervical cancer, the incidence of cervical cancer is still on the rise and its underlying pathogenesis remains unknown. Patients can be divided into 3 categories based on treatment: early, locally advanced, and advanced, which include stable, recurrent, or metastatic tumors. In addition to local or radical surgical treatment used to treat very early cervical cancer, radiotherapy and classical cytotoxic drugs are also widely used to treat patients with advanced cervical cancer [28]. Therefore, there is an urgent need to find reliable biomarkers and related drugs for diagnosis and prognosis of cervical cancer. In recent years, a large amount of microarray and sequencing data has been generated owing to the rapid development of bioinformatics, which provides comprehensive and convenient methods to screen genetic alterations and identify molecular mechanisms for the diagnosis, therapy, and prognosis of cancers.
In this study, GSE6791, GSE9750, GSE63514, and GSE67522 were analyzed for DEGs between cervical cancer tissues and normal tissues via GEO2R tools. A total of 78 DEGs (51 upregulated genes and 27 downregulated genes) were selected. Next, GO and KEGG analyses were performed using the DAVID website. In GO terms of Biological Process (BP) annotation, it was mainly enriched in the “mitotic nuclear divsion”, “cell proliferation”, “cell division”, and “DNA replication”. In Cellular Component (CC) annotation, it was enriched in the “nucleus”, “nucleoplasm”, “cytosol”, and “midbody”. In Molecular Function (MF) annotation, it was enriched in the “protein binding”, “protein kinase binding”, “single-stranded DNA binding”, and “ATP binding”. Additionally, in the KEGG pathway, it was enriched in “cell cycle” and “DNA replication”. Moreover, 78 DEGs were analyzed in the STRING database, and the top 10 hub genes (CDK1, CDC20, AURKA, TOP2A, ASPM, NCAPG, KIF23, CENPF, KIF20A, and PRC1) were selected using Cytoscape software.
A total 8 hub DEGs were enriched in the cell cycle-related pathways. The operation of the cell cycle is regulated by cyclin-dependent CDK kinases, and abnormal proliferation of the cell cycle can lead to carcinogenesis. Cyclin therapy is treatment of the cell cycle, and cyclin-dependent stimulated CDKs, Polo-like kinases, and WEE kinases are the main targets of cell cycle therapy drugs [29,30]. Cell cycle therapy can also activate tumor immunity, control metabolic function, and regulate transcription levels [31]. Patients’ responses to cell cycle therapy support the significance of cell cycle in the treatment of cervical cancer [32,33].
In the bioinformatics analysis, 10 hub genes were selected and verified based on TCGA. Finally, we identified 3 genes (CDK1, AURKA, and PRC1) as potential targets for predicting cervical cancer prognosis. High expression levels of CDK1 and PRC1 are associated with better clinical outcomes, while high expression of AURKA predicts poor survival in cervical cancer patients. The expression levels of these 3 genes were verified by immunostaining data from the HPA database. To confirm the important role of these 10 central genes in cervical cancer, mutation rates of these genes were further studied online in the cBioPortal. The results revealed that AURKA and PRC1 had higher mutation rates. Among the 3 central genes, only AURKA was positively correlated with clinical stage, while the other 2 had no statistically significant difference. Finally, based on the above analysis, AURKA is determined to be the key functional protein in these candidate genes.
AURKA is a serine/threonine kinase that plays an important regulatory role in cell mitosis. AURKA is overexpressed in several human malignancies and is the key to centrosome replication, maturation, and separation. Previous studies have shown that AURKA is elevated in most human cancers and is associated with adverse clinical outcomes in patients with lung cancer [34], liver cancer [35], and colorectal cancer [36]. A large number of studies have confirmed that AURKA, as an oncogene, affects many tumor biological processes, such as proliferation, genomic instability, aneuploid karyotype production, invasion, and metastasis. Xiaojiang et al [38] found that palmatine, an inhibitor of AURKA, can induce G2/M phase arrest and mitochondria-induced endogenous apoptosis of colon cancer cells by binding to AURKA. Silencing of AURKA resulted in the degradation of p27 and Bax in the cytoplasm, which decreased the viability of gastric cancer cells [39]. Shao et al reported that AURKA expression was significantly increased in tissues of cervical cancer patients. Further studies showed that hsa_circ_0075341 enhances the proliferation and invasion ability of cervical cancer cells by isolating miR-149-5p and upregulating AURKA expression [40]. Robert et al found that CDK1 activation initiated mitotic activation that AURKA was involved in. In addition, activated AURKA and PLK1 further react on CDK1 to promote mitosis and participate in spindle formation and centromere division in mitosis [41]. In the preset study, we found that the upregulation of AURKA was closely related to the survival prognosis and TNM clinical stage of cervical cancer, suggesting that AURKA is a potential target for clinical treatment of cervical cancer.
Using integrated bioinformatic analyses, AURKA was identified to play an important role in the development of cervical cancer, but few drugs designed to target this biomarker have been used to treat cervical cancer. In the present study, AURKA matched 21 FDA-approved drugs. Among the 21 drugs, the top 9 drugs – Fostamatinib, Regorafenib, Dasatinib, Brigatinib, Ponatinib, Pazopanib, Sorafenib, Imatinib, Nintedanib, and Acetylsalicylic acid (confidence score >0.3) – were analyzed. Fostamatinib is an oral tyrosine kinase inhibitor, which is involved in B cell receptor signaling and may be involved in regulating autoantibody production [42]. Regorafenib is a novel multikinase inhibitor that targets tumor genesis, tumor angiogenesis, and maintenance of signaling in the tumor microenvironment by inhibiting multiple protein kinases that promote tumor growth [43]. Dasatinib [44], Ponatinib [45], and ImatinibJournal [46] are tyrosine kinase inhibitors that effectively lock tyrosine kinase into inactive conformation by binding to highly conserved ATP binding sites, thereby inhibiting BCR-ABL activity. They can inhibit multiple oncogenic pathways activated by BCR-ABL tyrosine kinase and promote cell proliferation and survival, including the MAPK/ERK pathway, JAK-Stat pathway, and PI3K/Akt pathway. Brigatinib is an anaplastic lymphoma kinase inhibitor for the treatment of anaplastic lymphoma kinase-positive metastatic non-small cell lung cancer [47]. Pazopanib is a small-molecule inhibitor of a variety of protein tyrosine kinases, with potential antitumor activity [48]. Sorafenib works by stopping the progression of cancer and blocking the therapeutic replication of potentially malignant cells [49]. Nintedanib is a small-molecule kinase inhibitor that competitively inhibits both non-receptor tyrosine kinases and receptor tyrosine kinases, and these inhibitory effects increase fibroblast proliferation, migration, and transformation [50].
At present, the main drugs used for treatment for cervical cancer are Bevacizumab, Sunitinib, Olaparib, and cetuximab. In addition, Pazopanib and Nintedanib, obtained by our analysis, have also been widely used in the treatment of cervical cancer due to their anti-angiogenesis and tumor development inhibition [5,51]. However, with increased efficacy of these drugs, their adverse effects gradually increase. Therefore, we need to find more effective target genes and related drugs.
As mentioned above, the hub genes related to cervical cancer are mostly concentrated in the cell cycle pathway, and AURKA has been identified as a key gene affecting the occurrence and development of cervical cancer. Thus, we analyzed the mechanism of action of AURKA-related drugs in tumor therapy and found that these drugs are mostly targeted kinase inhibitors, which block tumor growth by affecting cell proliferation-related signaling pathways. Although the role of these drugs in the treatment of cervical cancer remains to be further determined, our results provide evidence of the important role of cell cycle signaling in the development of cervical cancer and once again support the possible efficacy of molecular-targeted therapies for cervical cancer.
Nevertheless, there exist some inevitable difficulties in this study that should be taken into consideration. For example, did not perform additional molecular and cellular biology experiments to verify their properties. However, our laboratory is currently carrying out relevant experiments to explore the role of AURKA in cervical cancer. The current analysis of samples collected from patients with cervical cancer is not statistically significant, so more samples need to be collected for verification. Although experimental data obtained from existing databases reduce confidence, the results of this study provide preliminary insights into the cell cycle signaling pathway involved in AURKA and its role in cervical cancer development, as well as AURKA gene-related targeting agents. This once again supports the use of cell cycle biologics and related potential drugs in the treatment of cervical cancer.
Conclusions
In summary, a total of 78 DEGs, including 51 upregulated genes and 27 downregulated genes, were identified in cervical cancer by integrated bioinformatic analysis. The further analysis identified that AURKA may account for the tumorigenesis and prognosis of cervical cancer. The potential drugs associated with AURKA gene were identified, which will provide new ideas for the treatment of cervical cancer. Although 4 data sets were been obtained and analyzed strictly and comprehensively, further experiments and molecular mechanism exploration are still needed to clarify how AURKA and its related hub genes affect cervical cancer proliferation. Our study provides a new direction for understanding the molecular mechanism and promoting clinical drug research for treatment of cervical cancer.
Figures
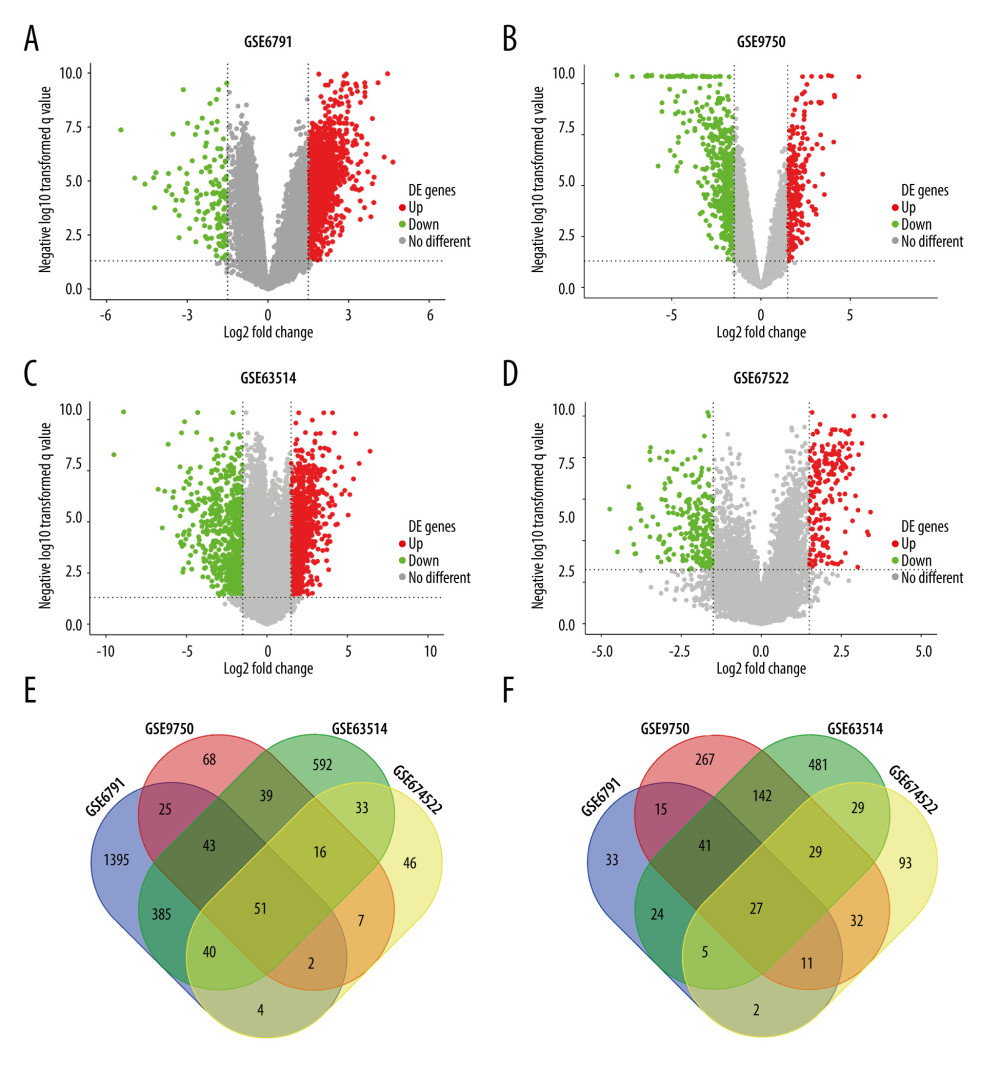 Figure 1. 78 overlapping DEGs among GSE6791, GSE9750, GSE63514, and GSE67522 datasets. The volcano plots of DEGs in (A) GSE6791, (B) GSE9750, (C) GSE63514, and (D) GSE67522 green dots and red dots represent the significantly downregulated and upregulated DEGs respectively. The upregulated (E) and downregulated (F) overlapping DEGs are illustrated in the Venn diagram. The relevant graphics are made through the Image GP (http://www.ehbio.com/Cloud_Platform/front/).
Figure 1. 78 overlapping DEGs among GSE6791, GSE9750, GSE63514, and GSE67522 datasets. The volcano plots of DEGs in (A) GSE6791, (B) GSE9750, (C) GSE63514, and (D) GSE67522 green dots and red dots represent the significantly downregulated and upregulated DEGs respectively. The upregulated (E) and downregulated (F) overlapping DEGs are illustrated in the Venn diagram. The relevant graphics are made through the Image GP (http://www.ehbio.com/Cloud_Platform/front/). 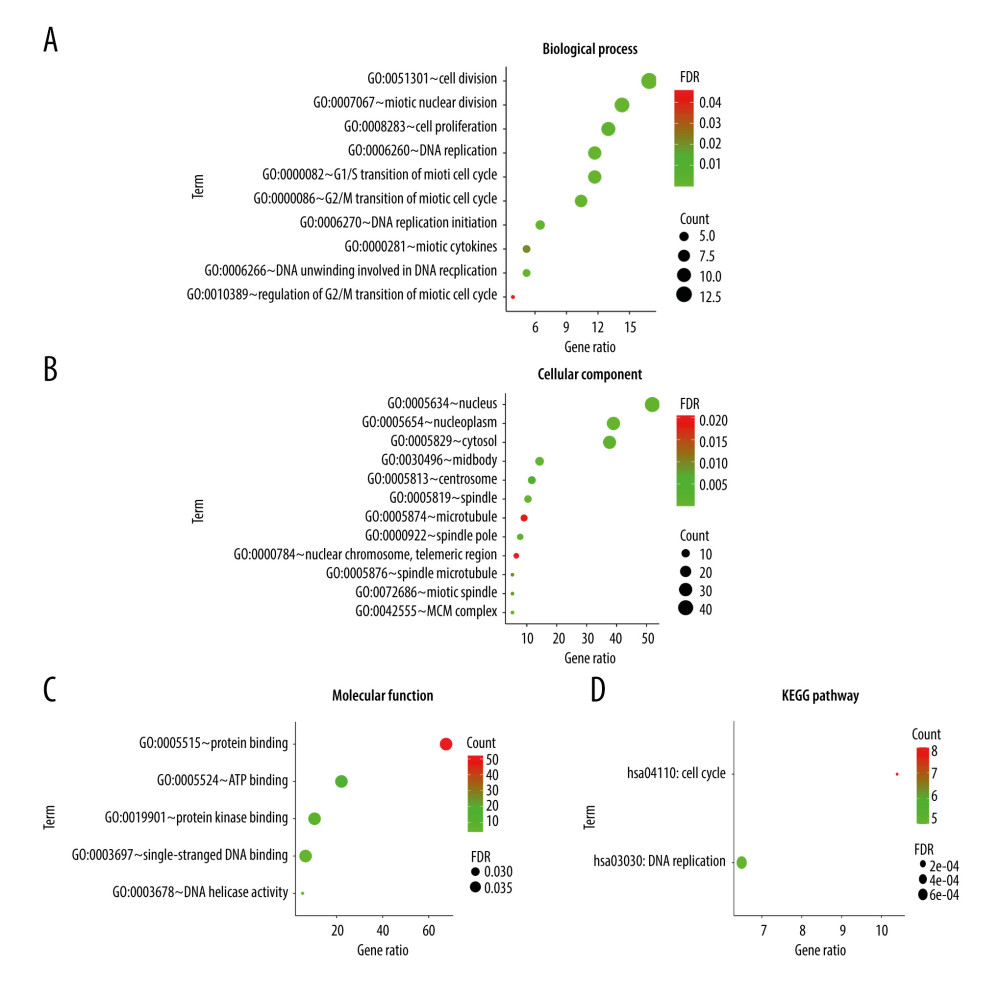 Figure 2. GO and KEGG enrichment analyses via the (https://david.ncifcrf.gov/) and the Image GP tool. The GO enrichment analysis of the overlapping DEGs in the categories of (A) biological process, (B) cellular component and (C) molecular functions. (D) The KEGG pathway enrichment analysis of the overlapping DEGs.
Figure 2. GO and KEGG enrichment analyses via the (https://david.ncifcrf.gov/) and the Image GP tool. The GO enrichment analysis of the overlapping DEGs in the categories of (A) biological process, (B) cellular component and (C) molecular functions. (D) The KEGG pathway enrichment analysis of the overlapping DEGs. 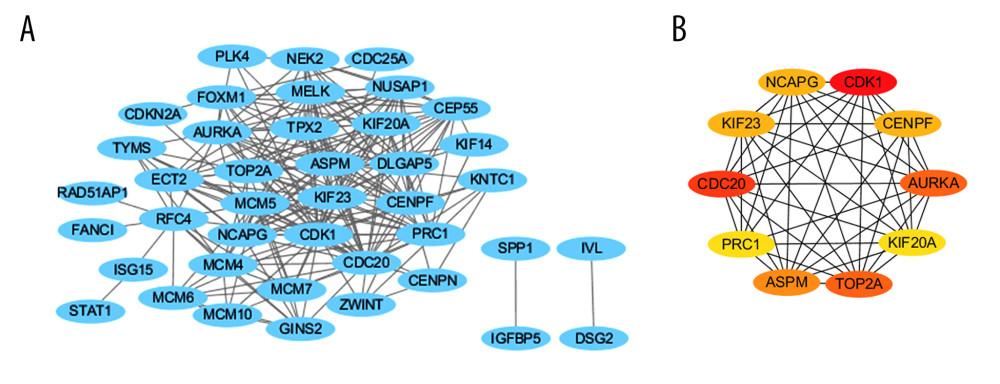 Figure 3. PPI network and hub gene identification via the cytoscape tool (http://www.cytoscape.org/). (A) PPI network of differentially expressed genes, including 41 nodes and 218 edges. The blue represents all the differentially expressed genes. All parameters in cytoHubba were set by default. (B) The top 10 genes in the PPI networks. The descending color from dark to light represents the weakening of hub gene interaction intensity.
Figure 3. PPI network and hub gene identification via the cytoscape tool (http://www.cytoscape.org/). (A) PPI network of differentially expressed genes, including 41 nodes and 218 edges. The blue represents all the differentially expressed genes. All parameters in cytoHubba were set by default. (B) The top 10 genes in the PPI networks. The descending color from dark to light represents the weakening of hub gene interaction intensity. 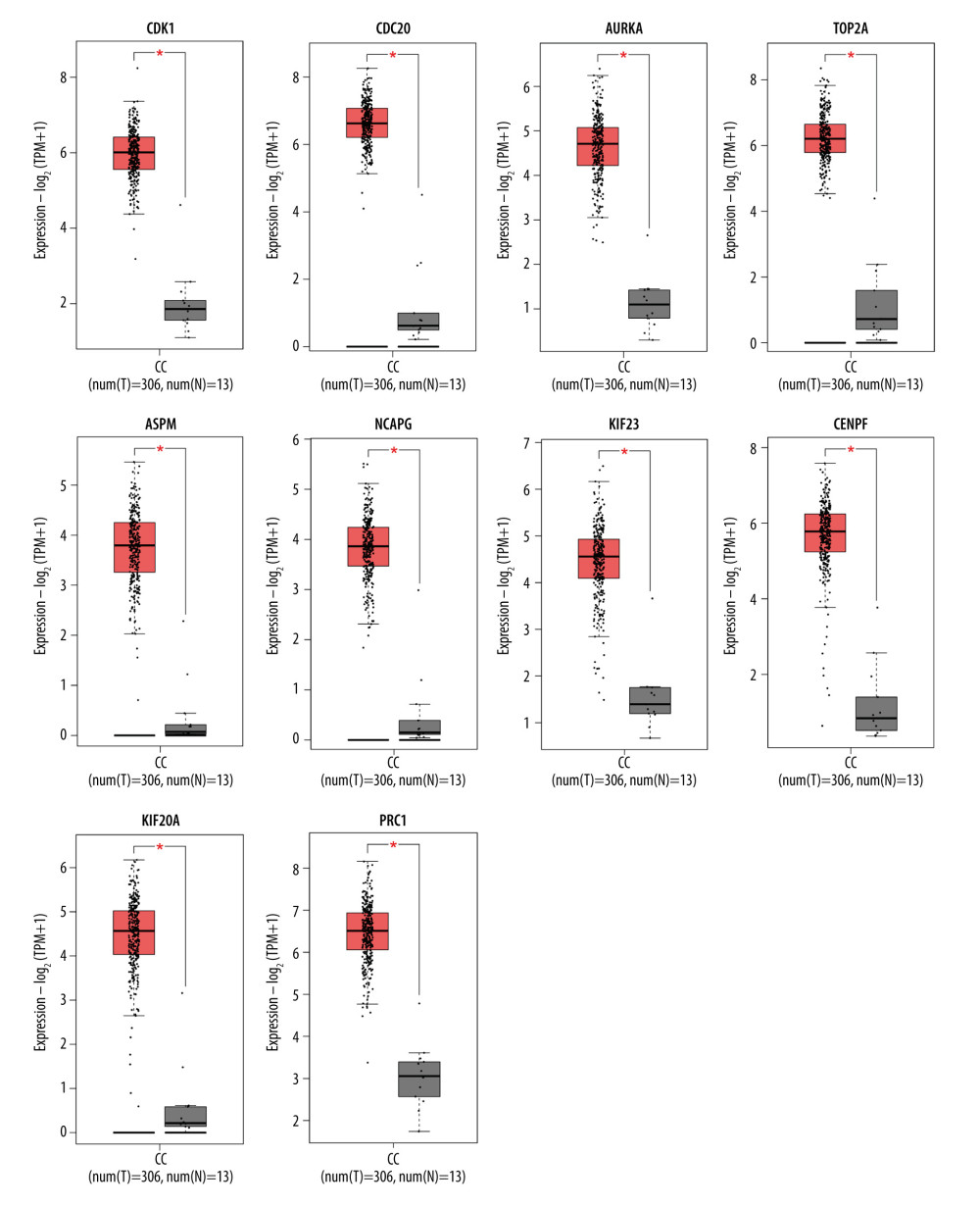 Figure 4. Boxplot graphs showing the expression levels of hub genes in normal cervical tissue and cervical cancer tissue via GEPIA website (http://gepia2.cancerpku.cn/). Red color represents tumor samples, while gray color represents normal samples. Red asterisks indicate P<0.05. CC – cervial cancer.
Figure 4. Boxplot graphs showing the expression levels of hub genes in normal cervical tissue and cervical cancer tissue via GEPIA website (http://gepia2.cancerpku.cn/). Red color represents tumor samples, while gray color represents normal samples. Red asterisks indicate P<0.05. CC – cervial cancer. 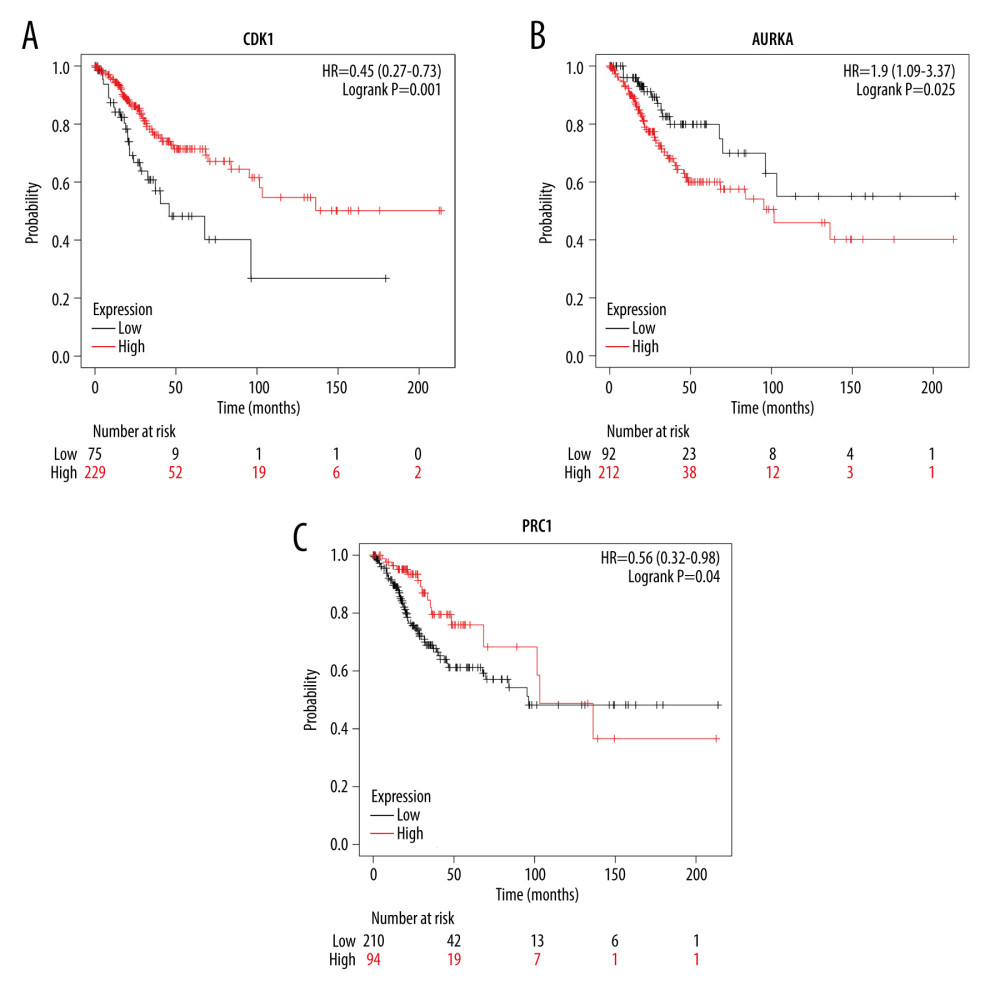 Figure 5. Overall survival analysis of hub genes using the Kaplan-Meier curves (https://kmplot.com/analysis/index.php?p=background). The association between the expression levels of CDK1 (A), AURKA (B), and PRC1 (C) and overall survival rate of the patients with cervical cancer was analysed by using the KM plotter.
Figure 5. Overall survival analysis of hub genes using the Kaplan-Meier curves (https://kmplot.com/analysis/index.php?p=background). The association between the expression levels of CDK1 (A), AURKA (B), and PRC1 (C) and overall survival rate of the patients with cervical cancer was analysed by using the KM plotter. 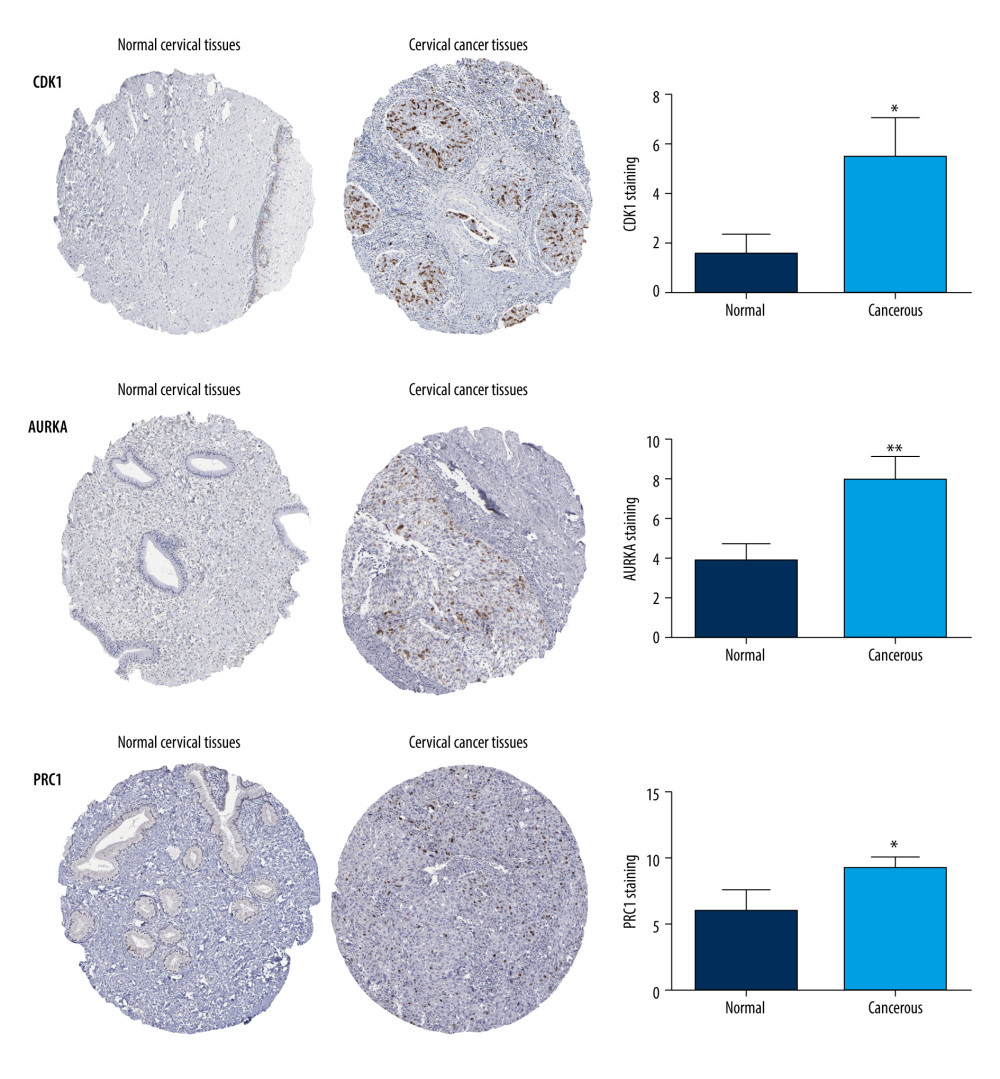 Figure 6. Protein expression levels of hub genes (CDK1, AURKA and PRC1) were analysed using the Human Protein Atlas database (https://www.proteinatlas.org/). The number of staining cells was less than 25%. * P<0.05 and ** P<0.01.
Figure 6. Protein expression levels of hub genes (CDK1, AURKA and PRC1) were analysed using the Human Protein Atlas database (https://www.proteinatlas.org/). The number of staining cells was less than 25%. * P<0.05 and ** P<0.01. 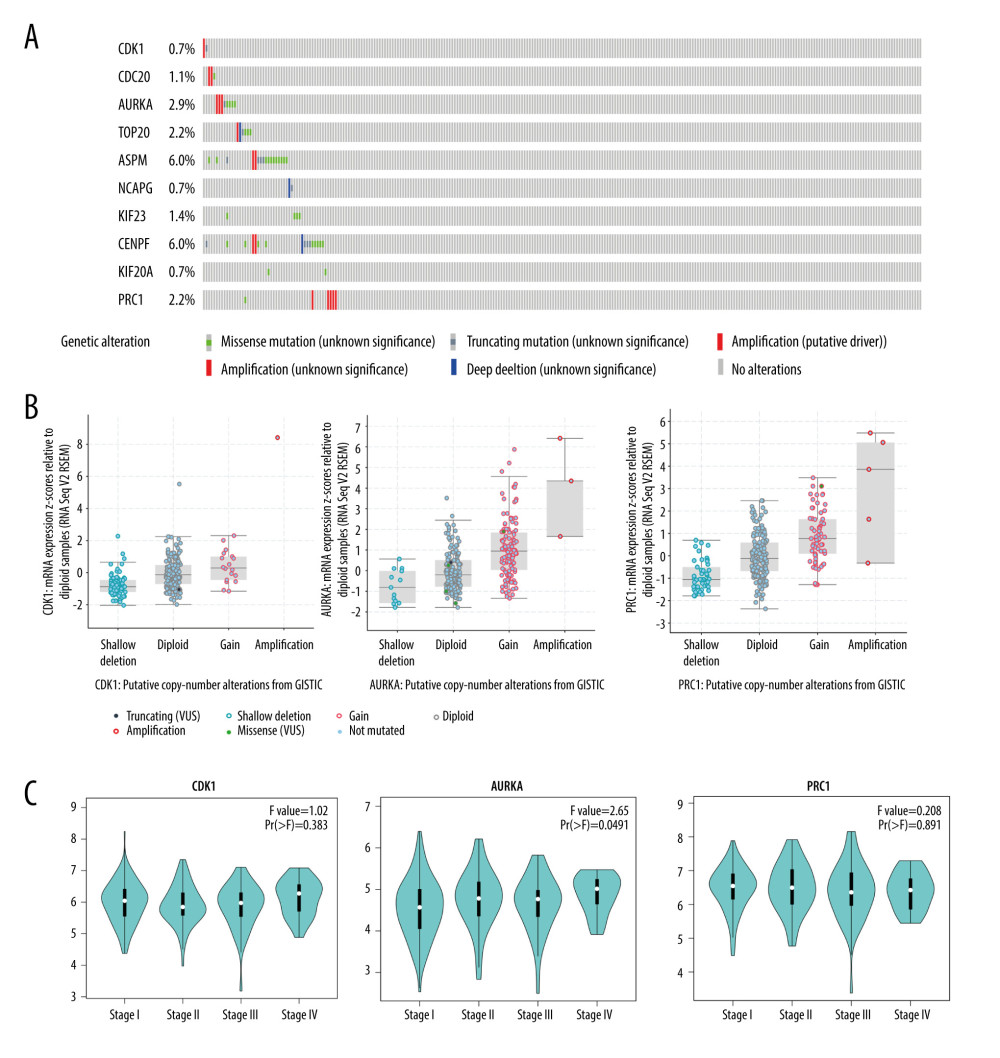 Figure 7. Validation of the differential expression of 10 hub genes’ mutation rates and various clinical stages. (A) The cBioPortal database (http://www.cbioportal.org) shows that the 10 hub genes are known to be mutated in cervial cancer. (B) The mutation status of CDK1, AURKA, and PRC1 in cervial cancer is mainly shallow deletion, and the mutation status of AURKA and PRC1 is mainly amplification. (C) In the pathological staging of cervial cancer, the expression of CDK1, AURKA, and PRC1 were significantly differentiated in various clinical stages.
Figure 7. Validation of the differential expression of 10 hub genes’ mutation rates and various clinical stages. (A) The cBioPortal database (http://www.cbioportal.org) shows that the 10 hub genes are known to be mutated in cervial cancer. (B) The mutation status of CDK1, AURKA, and PRC1 in cervial cancer is mainly shallow deletion, and the mutation status of AURKA and PRC1 is mainly amplification. (C) In the pathological staging of cervial cancer, the expression of CDK1, AURKA, and PRC1 were significantly differentiated in various clinical stages. References
1. de Martel C, Georges D, Bray F, Global burden of cancer attributable to infections in 2018: A worldwide incidence analysis: Lancet Global Health, 2020; 8(2); E180-90
2. Canfell K, Kim JJ, Brisson M, Mortality impact of achieving WHO cervical cancer elimination targets: A comparative modelling analysis in 78 low-income and lower-middle-income countries: Lancet, 2020; 395(10224); 591-603
3. Yuan Y, Cai XS, Shen FR, Ma F, HPV post-infection microenvironment and cervical cancer: Cancer Lett, 2021; 497; 243-54
4. Wang R, Pan W, Jin L, Human papillomavirus vaccine against cervical cancer: Opportunity and challenge: Cancer Lett, 2020; 471; 88-102
5. Cohen AC, Roane BM, Leath CA, Novel therapeutics for recurrent cervical cancer: Moving towards personalized therapy: Drugs, 2020; 80(3); 217-27
6. Chopra S, Gupta M, Mathew A, Locally advanced cervical cancer: A study of 5-year outcomes: Indian J Cancer, 2018; 55(1); 45-49
7. Armstrong AJ, Li XT, Tucker M, Molecular medicine tumor board: whole-genome sequencing to inform on personalized medicine for a man with advanced prostate cancer: Prostate Cancer Prostatic Dis, 2021; 24; 786-93
8. Huang JW, Deng XN, Wang YL, Analysis of copy number variations by low-depth whole-genome sequencing in fetuses with congenital cardiovascular malformations: Cytogenet Genome Res, 2020; 160; 643-49
9. Kondelin J, Martin S, Katainen R, No evidence of EMAST in whole genome sequencing data from 248 colorectal cancers: Genes Chromosomes Cancer, 2021; 60; 463-73
10. Bai JW, Shi JX, Li CZ, Whole genome sequencing of skull-base chordoma reveals genomic alterations associated with recurrence and chordoma-specific survival: Nat Commun, 2021; 12(1); 757
11. Ward ZJ, Grover S, Scott AM, The role and contribution of treatment and imaging modalities in global cervical cancer management: Survival estimates from a simulation-based analysis: Lancet Oncol, 2020; 21(8); 1089-98
12. Wei J, Wang Y, Shi K, Wang Y, Identification of core prognosis-related candidate genes in cervical cancer via integrated bioinformatical analysis: Biomed Res Int, 2020; 2020; 8959210
13. Xue H, Sun ZJ, Wu WQ, Identification of hub genes as potential prognostic biomarkers in cervical cancer using comprehensive bioinformatics analysis and validation studies: Cancer Manag Res, 2021; 13; 117-31
14. Cai LY, Hu C, Yu SS, Identification and validation of a six-gene signature associated with glycolysis to predict the prognosis of patients with cervical cancer: Bmc Cancer, 2020; 20(1); 1133
15. Yang X, Li Q, The mechanism of CSRP2BP in cervial cancer: Eur J Immunol, 2019; 49; 1560-60
16. Zhao J, Cao HL, Zhang WF, SOX14 hypermethylation as a tumour biomarker in cervical cancer: Bmc Cancer, 2021; 21(1); 675
17. , The Gene Ontology Resource: 20 years and still GOing strong: Nucleic Acids Res, 2019; 47; D330-38
18. Kanehisa M, Furumichi M, Sato Y, KEGG: Integrating viruses and cellular organisms: Nucleic Acids Res, 2021; 49(D1); D545-51
19. Huang DW, Sherman BT, Tan Q, DAVID Bioinformatics Resources: Expanded annotation database and novel algorithms to better extract biology from large gene lists: Nucleic Acids Res, 2007; 35; W169-75
20. Szklarczyk D, Gable AL, Lyon D, STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets: Nucleic Acids Res, 2019; 47(D1); D607-13
21. Elahi A, Babamir SM, Identification of protein complexes based on core-attachment structure and combination of centrality measures and biological properties in PPI weighted networks: Protein J, 2020; 39(6); 681-702
22. Tang Z, Kang B, Li C, GEPIA2: An enhanced web server for large-scale expression profiling and interactive analysis: Nucleic Acids Res, 2019; 47(W1); W556-60
23. Ranstam J, Cook JA, Kaplan-Meier curve: Br J Surg, 2017; 104(4); 442
24. Digre A, Lindskog C, The Human Protein Atlas-Spatial localization of the human proteome in health and disease: Protein Sci, 2021; 30(1); 218-33
25. Cerami E, Gao J, Dogrusoz U, The cBio Cancer Genomics Portal: An open platform for exploring multidimensional cancer genomics data (vol 2, pg 401, 2012): Cancer Discovery, 2012; 2(10); 960
26. Li HC, Pei F, Taylor DL, Bahar I, QuartataWeb: Integrated chemical-protein-pathway mapping for polypharmacology and chemogenomics: Bioinformatics, 2020; 36(12); 3935-37
27. Shrestha AD, Neupane D, Vedsted P, Kallestrup P, Cervical cancer prevalence, incidence and mortality in low and middle income countries: A systematic review: Asian Pac J Cancer Prev, 2018; 19(2); 319-24
28. Liu Y, An Q, Zhao X, A pan-HER-targeted approach for recurrent or late-stage cervical cancer therapy: Mechanisms, recent advances, and clinical prospects: Eur Rev Med Pharmacol Sci, 2020; 24(8); 4123-31
29. Ingham M, Schwartz GK, Cell-cycle therapeutics come of age: J Clin Oncol, 2017; 35(25); 2949-59
30. Wang X, Deng K, Wang C, Novel CDKs inhibitors for the treatment of solid tumour by simultaneously regulating the cell cycle and transcription control: J Enzyme Inhib Med Chem, 2020; 35(1); 414-23
31. Li J, Stanger BZ, Cell cycle regulation meets tumor immunosuppression: Trends Immunol, 2020; 41(10); 859-63
32. Mondala PK, Vora AA, Zhou T, Selective antisense oligonucleotide inhibition of human IRF4 prevents malignant myeloma regeneration via cell cycle disruption: Cell Stem Cell, 2021; 28(4); 623-36e9
33. Zhou S, Luo Q, Tan X, Erchen decoction plus huiyanzhuyu decoction inhibits the cell cycle, migration and invasion and induces the apoptosis of laryngeal squamous cell carcinoma cells: J Ethnopharmacol, 2020; 256; 112638
34. Woo JK, Kang JH, Shin D, Daurinol enhances the efficacy of radiotherapy in lung cancer via suppression of Aurora kinase A/B expression: Mol Cancer Ther, 2015; 14(7); 1693-704
35. Jin X, Wang J, Zou S, Cinobufagin triggers defects in spindle formation and cap-dependent translation in liver cancer cells by inhibiting the AURKA-mTOR-eIF4E Axis: Am J Chin Med, 2020; 48(3); 651-78
36. Shan B, Zhao R, Zhou J, AURKA increase the chemosensitivity of colon cancer cells to oxaliplatin by inhibiting the TP53-mediated DNA damage response genes: Biomed Res Int, 2020; 2020; 8916729
37. Carvalhal S, Ribeiro SA, Arocena M, The nucleoporin ALADIN regulates Aurora A localization to ensure robust mitotic spindle formation: Molecular biology of the cell, 2015; 26(19); 3424-38
38. Liu X, Zhang Y, Wu S, Palmatine induces G2/M phase arrest and mitochondrial-associated pathway apoptosis in colon cancer cells by targeting AURKA: Biochem Pharmacol, 2020; 175; 113933
39. Hou D, Che Z, Chen P, Suppression of AURKA alleviates p27 inhibition on Bax cleavage and induces more intensive apoptosis in gastric cancer: Cell Death Dis, 2018; 9(8); 781
40. Shao SQ, Wang C, Wang SL, Hsa_circ_0075341 is up-regulated and exerts oncogenic properties by sponging miR-149-5p in cervical cancer: Biomed Pharmacother, 2020; 121; 109582
41. Van Horn RD, Chu SY, Fan L, Cdk1 activity is required for mitotic activation of Aurora A during G(2)/M transition of human cells: J Biol Chem, 2010; 285(28); 21849-57
42. Moore DC, Gebru T, Muslimani A, Fostamatinib for the treatment of immune thrombocytopenia in adults: Am J Health Syst Pharm, 2019; 76(11); 789-94
43. Zhang N, Zhang S, Wu W, Regorafenib inhibits migration, invasion, and vasculogenic mimicry of hepatocellular carcinoma via targeting ID1-mediated EMT: Mol Carcinog, 2021; 60(2); 151-63
44. Alkebsi L, Wang X, Ohkawara H, Dasatinib induces endothelial-to-mesenchymal transition in human vascular-endothelial cells: Counteracted by cotreatment with bosutinib: Int J Hematol, 2021; 113(3); 441-55
45. Kidoguchi K, Ureshino H, Kizuka-Sano H, Efficacy and safety of ponatinib for patients with Philadelphia chromosome-positive acute lymphoblastic leukemia: A case series from a single institute: Int J Hematol, 2021; 114(2); 199-204
46. Willig JB, Vianna DRB, Beckenkamp A, Imatinib mesylate affects extracellular ATP catabolism and expression of NTPDases in a chronic myeloid leukemia cell line: Purinergic Signal, 2020; 16(1); 29-40
47. Nishio M, Yoshida T, Kumagai T, Brigatinib in Japanese patients with ALK-positive NSCLC previously treated with alectinib and other tyrosine kinase inhibitors: Outcomes of the phase 2 J-ALTA Trial: J Thorac Oncol, 2021; 16(3); 452-63
48. Er MM, Araz M, Karabacak M, Pazopanib-associated secondary adrenal insufficiency in a patient with malignant solitary fibrous tumor: J Oncol Pharm Pract, 2021; 27(8); 2049-52
49. Cao Y, Sun T, Guo X, Sorafenib versus apatinib both combined transarterial chemoembolization for hepatocellular carcinoma with portal vein tumor thrombosis: A comparative retrospective study: Front Oncol, 2021; 11; 673378
50. Kouroki M, Inagaki J, Fujisaki T, Nintedanib for treatment of progressive interstitial lung disease after peripheral blood stem cell transplantation in a patient with recurrent acute lymphoblastic leukemia: Pediatr Blood Cancer, 2021; 68(11); e29211
51. Luvero D, Plotti F, Lopez S, Antiangiogenics and immunotherapies in cervical cancer: An update and future’s view: Med Oncol, 2017; 34(6); 115
Figures
Figure 1. 78 overlapping DEGs among GSE6791, GSE9750, GSE63514, and GSE67522 datasets. The volcano plots of DEGs in (A) GSE6791, (B) GSE9750, (C) GSE63514, and (D) GSE67522 green dots and red dots represent the significantly downregulated and upregulated DEGs respectively. The upregulated (E) and downregulated (F) overlapping DEGs are illustrated in the Venn diagram. The relevant graphics are made through the Image GP (http://www.ehbio.com/Cloud_Platform/front/).Figure 2. GO and KEGG enrichment analyses via the (https://david.ncifcrf.gov/) and the Image GP tool. The GO enrichment analysis of the overlapping DEGs in the categories of (A) biological process, (B) cellular component and (C) molecular functions. (D) The KEGG pathway enrichment analysis of the overlapping DEGs.Figure 3. PPI network and hub gene identification via the cytoscape tool (http://www.cytoscape.org/). (A) PPI network of differentially expressed genes, including 41 nodes and 218 edges. The blue represents all the differentially expressed genes. All parameters in cytoHubba were set by default. (B) The top 10 genes in the PPI networks. The descending color from dark to light represents the weakening of hub gene interaction intensity.Figure 4. Boxplot graphs showing the expression levels of hub genes in normal cervical tissue and cervical cancer tissue via GEPIA website (http://gepia2.cancerpku.cn/). Red color represents tumor samples, while gray color represents normal samples. Red asterisks indicate P<0.05. CC – cervial cancer.Figure 5. Overall survival analysis of hub genes using the Kaplan-Meier curves (https://kmplot.com/analysis/index.php?p=background). The association between the expression levels of CDK1 (A), AURKA (B), and PRC1 (C) and overall survival rate of the patients with cervical cancer was analysed by using the KM plotter.Figure 6. Protein expression levels of hub genes (CDK1, AURKA and PRC1) were analysed using the Human Protein Atlas database (https://www.proteinatlas.org/). The number of staining cells was less than 25%. * P<0.05 and ** P<0.01.Figure 7. Validation of the differential expression of 10 hub genes’ mutation rates and various clinical stages. (A) The cBioPortal database (http://www.cbioportal.org) shows that the 10 hub genes are known to be mutated in cervial cancer. (B) The mutation status of CDK1, AURKA, and PRC1 in cervial cancer is mainly shallow deletion, and the mutation status of AURKA and PRC1 is mainly amplification. (C) In the pathological staging of cervial cancer, the expression of CDK1, AURKA, and PRC1 were significantly differentiated in various clinical stages. Tables
 Table 1. All 78 commonly differentially expressed genes (DEGs) were detected from four profile datasets, including 51 up-regulated genes and 27 down-regulated genes in the cervical cancer compared to normal cervical tissues.
Table 1. All 78 commonly differentially expressed genes (DEGs) were detected from four profile datasets, including 51 up-regulated genes and 27 down-regulated genes in the cervical cancer compared to normal cervical tissues.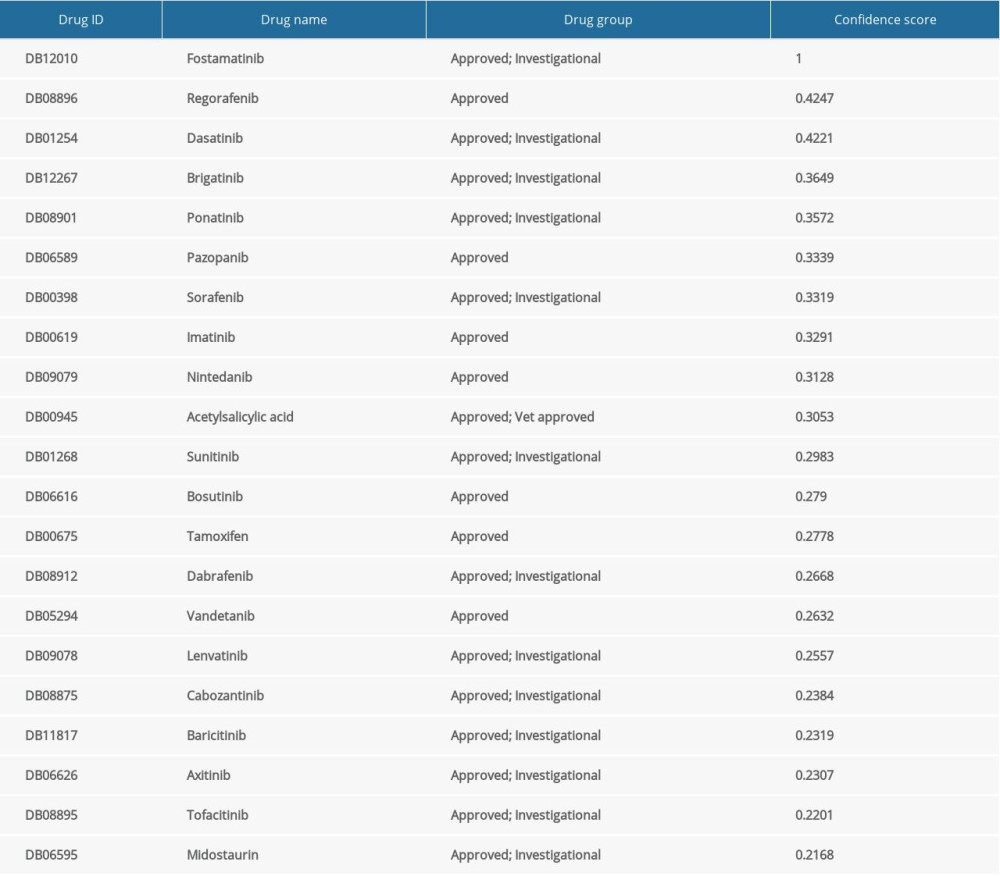 Table 2. The significant drugs targeted to AURKA.Table 1. All 78 commonly differentially expressed genes (DEGs) were detected from four profile datasets, including 51 up-regulated genes and 27 down-regulated genes in the cervical cancer compared to normal cervical tissues.Table 2. The significant drugs targeted to AURKA.
Table 2. The significant drugs targeted to AURKA.Table 1. All 78 commonly differentially expressed genes (DEGs) were detected from four profile datasets, including 51 up-regulated genes and 27 down-regulated genes in the cervical cancer compared to normal cervical tissues.Table 2. The significant drugs targeted to AURKA. In Press
Clinical Research
Analysis of the Clinical Characteristics and Endoscopic Features of Phytobezoar-Induced Ulcers and Gastric ...Med Sci Monit In Press; DOI: 10.12659/MSM.952191
Clinical Research
Effect of Indirect Co-Culture With Gingival Mesenchymal Stem Cells on Cytokine Secretion in Primary Oral Sq...Med Sci Monit In Press; DOI: 10.12659/MSM.952439
Clinical Research
Comparison of Sleep Architecture in Individuals Aged 65 to 80 Years With and Without Mild Cognitive Impairm...Med Sci Monit In Press; DOI: 10.12659/MSM.952493
Clinical Research
Effects of Single-Bout Endurance Exercise Intensity on Peripheral Neurotrophic Factors in Patients With Isc...Med Sci Monit In Press; DOI: 10.12659/MSM.952089
Most Viewed Current Articles
17 Jan 2024 : Review article 14,176,514
Vaccination Guidelines for Pregnant Women: Addressing COVID-19 and the Omicron VariantDOI :10.12659/MSM.942799
Med Sci Monit 2024; 30:e942799
13 Nov 2021 : Clinical Research 3,760,677
Acceptance of COVID-19 Vaccination and Its Associated Factors Among Cancer Patients Attending the Oncology ...DOI :10.12659/MSM.932788
Med Sci Monit 2021; 27:e932788
14 Dec 2022 : Clinical Research 2,466,264
Prevalence and Variability of Allergen-Specific Immunoglobulin E in Patients with Elevated Tryptase LevelsDOI :10.12659/MSM.937990
Med Sci Monit 2022; 28:e937990
16 May 2023 : Clinical Research 708,906
Electrophysiological Testing for an Auditory Processing Disorder and Reading Performance in 54 School Stude...DOI :10.12659/MSM.940387
Med Sci Monit 2023; 29:e940387