22 October 2024: Review Articles
Targeting MuRF1 to Combat Skeletal Muscle Wasting in Cardiac Cachexia: Mechanisms and Therapeutic Prospects
Xiaotong Liu1E, Ya Wen2E, Yanmei LuDOI: 10.12659/MSM.945211
Med Sci Monit 2024; 30:e945211






Abstract
ABSTRACT: Cardiac cachexia, the terminal stage of chronic heart failure, is characterized by severe systemic metabolic imbalances and significant weight loss, primarily resulting from skeletal muscle mass depletion. Despite the detrimental consequences, there is no standardized and clinically-approved intervention currently available for cardiac cachexia. In the context of cardiac cachexia, accelerated protein turnover, that is, inhibited protein synthesis and enhanced protein degradation, plays a crucial role in skeletal muscle wasting. This process is primarily mediated by various proteins encoded by atrogenes. Among them, the atrogene Trim63 (tripartite motif family 63) and its encoded protein MuRF1 have been extensively studied. This review article aims to elucidate the pathogenic mechanisms underlying skeletal muscle wasting in cardiac cachexia, describe the biochemical characteristics of MuRF1, and provide an overview of the investigation into MuRF1-targeting inhibitors. The ultimate goal is to offer novel strategies for the clinical treatment for skeletal muscle wasting associated with cardiac cachexia.
Keywords: Heart Failure, Cachexia, TRIM63 protein, human, Atrophy, Muscles, Humans, Tripartite Motif Proteins, Ubiquitin-Protein Ligases, Muscle, Skeletal, Muscle Proteins, Muscular Atrophy, Animals
Introduction
Chronic heart failure (CHF) is a multifactorial clinical syndrome characterized by structural or functional abnormalities of the heart [1]. In its terminal stage, CHF can lead to cardiac cachexia, a condition exacerbated by multi-organ dysfunction and disordered metabolism. The term “cachexia” is derived from the Greek “
The clinical manifestations of cardiac cachexia include severe edema, weight loss of at least 5% irrespective of initial body weight, or a body mass index (BMI) of less than 20 kg/m2 over a period of 12 months. Patients may also experience fatigue, reduced muscle mass and strength, dyspnea, anorexia, anemia, and abnormal blood biomarker levels such as elevated CRP (C-reactive protein) (>5.0 mg/l), increased IL-6 (interleukin-6) (>4.0 pg/ml), low hemoglobin (<12 g/dl), and/or decreased serum albumin (<3.2 g/dl) [4–6] (Figure 1). In developed countries and regions like the United States, Europe, and Japan, the annual mortality rate for patients with cardiac cachexia is 20–40% [3]. Research by Anker et al demonstrated a stark contrast in mortality rates: 17% for heart failure patient without cachexia versus 50% for those with cachexia after an 18-month follow-up. Their study, employing a Cox proportional hazards model for survival analysis, further corroborated cardiac cachexia as an independent predictor of mortality in CHF patients [7].
Cachexia often occurs secondary to severe conditions such as sepsis, severe trauma, and many chronic systemic inflammatory diseases, such as chronic obstructive pulmonary disease (COPD), CHF, chronic renal failure, cancer, type 2 diabetics, and rheumatoid arthritis [8]. In cachexia, the rate of catabolism exceeds anabolism, leading primarily to a reduction in body mass dominated by skeletal muscle wasting [9]. Skeletal muscle wasting is characterized by a decline in muscle mass and dysfunction. Patients with cachexia frequently experience a reduced capacity for exercise, disability, weakness, and fatigue, which adversely affect their quality of life and increase the risk of mortality and healthcare costs [10–13]. Preserving skeletal muscle mass and function when healthy may improve outcomes for patients with cardiac cachexia. While research continues into various nutritional supplements and medications to prevent muscle wasting associated with diseases, exercise training remains the only clinically-approved intervention that is both safe and effective. However, it is not recommended for patients with severely limited capacity for exercise, compromised heart function, or those who are bedridden or immobilized for extended periods [8,14,15].
Skeletal muscle atrophy primarily results from an imbalance between protein synthesis and degradation. The ubiquitin-proteasome-system (UPS), autophagy-lysosome system, and apoptosis are implicated in increased protein breakdown, with UPS playing a predominant role in the disassembly of myofibrils [8,10,16,17]. Bodine et al identified 2 critical atrophy-related genes, Trim63 and Fbxo32, which are overexpressed in striated muscle under atrophic conditions and encode for Muscle RING-Finger 1 (MuRF1) and MAFbx/atrogin-1, respectively [18]. These genes are upregulated in various atrophy scenarios, including fasting [19,20], exposure to glucocorticoids [21,22], denervation [23,24], hindlimb suspension [25], and immobilization [26,27]. The transcription of atrogenes Trim63 and Fbxo32 is under the regulation of NF-κB, Foxo, and SMAD signaling pathways, with several inhibitors of these pathways demonstrated to indirectly reduce MuRF1 and MAFbx expression [28–30].
Evidence has mounted that MAFbx plays a role in the myocardial hypertrophy triggered by the NF-κB pathway [31]. As a result, suppressing MAFbx activity may alleviate the symptoms of pathological hypertrophy and could even help in reversing the course of heart failure. Nonetheless, it remains a significant gap in the field that no studies have yet announced the development of small-molecule inhibitors designed to specifically target and suppress MAFbx. Therefore, this article emphasizes the introduction of MuRF1-targeting inhibitors. Recent research involving MuRF1-targeting inhibitors, such as MyoMed-205, has shown promise in mitigating skeletal muscle wasting and partially reversing myocardial fibrosis in a rat model of heart failure with preserved ejection fraction (HFpEF) over a 32-week period. Targeting these pathways could therefore be beneficial for treating skeletal muscle wasting associated with cardiac cachexia [32].
Pathophysiological Mechanism of Cardiac Cachexia
Patients with CHF experience a cascade of pathophysiological changes, including significant fluctuations in neurohormonal, metabolic hormone levels, and heightened inflammatory responses. These alterations precipitate a catabolic state, characterized by reduced muscle and adipose tissue mass, bone density, and mineral content. Concurrently, these patients often have loss of appetite, increased feelings of fullness, and gastrointestinal dysfunction. This persistent catabolic and depletion state culminates in cardiac cachexia, which clinically manifests as frailty, fatigue, weight loss, and diminished physical strength (Figure 2).
Alterations in Hormone Levels
ANG-II (ANGIOTENSIN-II):
The enhanced release of Ang-II, a primary component of the renin–angiotensin–aldosterone system (RAAS), occurs in response to increased sympathetic activity. This elevation contributes to muscle wasting, diminished appetite, and weight loss upon receptor engagement [33]. Ang-II orchestrates the balance of protein synthesis and degradation by boosting NADPH oxidase activity, resulting in elevated reactive oxygen species (ROS) production [33]. Its influence extends to the hypothalamic appetite regulation centers, where it modulates the secretion of neuropeptide-Y (NPY), orexin, and corticotropin-releasing hormone (CRH), thereby reducing appetite [35].
GHRELIN-GROWTH HORMONE (GH)-AXIS:
Ghrelin, a natural agonist for the growth hormone secretagogue receptor (GHS-R) [36], is found in higher concentrations in patients with cardiac cachexia compared to those with CHF but without cachexia [37]. This hormone stimulates secretion of growth hormone (GH), which predominantly promotes muscle growth through the insulin-like growth factor-1 (IGF-1) signaling pathway [38]. However, research indicates that patients with CHF may develop ghrelin resistance due to elevated levels of this “hunger hormone” during catabolic states, alongside GH resistance, which is characterized by increased GH levels but decreased IGF-1 [36,39]. This hormonal imbalance is associated with loss of muscle mass and contributes to progression of cardiac cachexia in CHF patients.
ADIPOKINES – ADIPONECTIN AND LEPTIN:
Abnormal lipid and glucose metabolism may stem from increased levels of adiponectin and a relatively higher concentration of leptin compared to healthy individuals – likely due to weight loss and reduced adipose tissue [40–42]. Doehner et al documented that heightened leptin levels in CHF patients, irrespective of cachexia status, can contribute to decreased insulin sensitivity and development of insulin resistance [43,44]. Moreover, this increase in leptin can also amplify feelings of fullness, diminish appetite, and reduce nutrient intake in individuals with cardiac cachexia [45].
TESTOSTERONE:
The catabolic state in cardiac cachexia is marked by diminished testosterone and elevated cortisol levels, which further intensify muscle wasting, inflammatory responses, and oxidative stress [5,46,47].
Excessive Inflammatory Responses
In the setting of cardiac cachexia, levels of inflammatory mediators, particularly TNF-α, IL-1 and IL-6, are significantly elevated [48]. These cytokines are predominantly released by monocytes and macrophages. Additionally, cardiomyocytes contribute to cytokine release due to hypoxic conditions induced by CHF, which serves as a primary pathway for release of these key inflammatory agents [49]. An increase in neurohormonal secretion, observed in CHF patients both with and without cardiac cachexia, further augments the release of inflammatory factors, including cortisol, catecholamines, and RAAS-related hormones such as aldosterone and Ang-II [48,50]. Notably, TNF-α concentrations are reported to be significantly higher in patients with cardiac cachexia compared to those without [51], and research by Anker et al has identified an association between TNF-α levels and weight loss in these patients [52]. Previous studies have demonstrated that TNF-α, IL-1, and IL-6 are critical upstream triggers for UPS activity, leading to increased protein degradation and skeletal muscle wasting [48]. Furthermore, the adverse effects of TNF-α on vascular endothelial cell function and capillary density reduction impede blood supply to the skeletal muscles, thereby disrupting energy metabolism and nutritional status, and exacerbating the progression of cardiac cachexia [5,40,53].
Unbalanced Protein Synthesis and Degradation
INHIBITION OF PROTEIN SYNTHESIS:
Both IGF-1 and insulin stimulate protein synthesis via the PI3K-Akt-mTOR pathway. However, in the catabolic state characteristic of CHF, protein synthesis is impeded due to diminished levels of circulating IGF-1 and prevalent insulin resistance in patients [38,54,55]. Ang-II exacerbates this condition by inhibiting the release of IGF-1 into the bloodstream, further hindering protein synthesis, as noted by Sukhanov [56]. Moreover, Breitbart et al found that myostatin, a potent inhibitor of muscle growth, is secreted into the circulation by the heart during failure, playing a pivotal role in the muscle wasting seen in cardiac cachexia [57]. Morissette et al reported that the inhibitory effect of myostatin on protein synthesis could be due to its interference with IGF-1-mediated phosphorylation of Akt, also known as protein kinase B, as a result of increased myostatin level [58].
ACCELERATION OF PROTEIN DEGRADATION:
In vertebrates, protein catabolism is predominantly facilitated by 3 key systems: the UPS, the autophagic lysosomal system, and the caspase-dependent apoptotic system.
UBIQUITIN-PROTEASOME SYSTEM (UPS):
UPS is hyperactivated by atrophic stimuli, leading to breakdown of key skeletal muscle proteins, including myosin, myosin binding protein C (MyBP-C), and α-actin [59]. The ubiquitination process involves a series of enzyme-catalyzed reactions in which the ubiquitin-activating enzyme (E1) forms high-energy thioester bonds between cysteine residues of E1 and the C-terminal glycine residues of ubiquitin. This activated ubiquitin is then conveyed to the ubiquitin-conjugating enzyme (E2), creating an E2-ubiquitin complex. Following this, the ubiquitin ligase (E3) facilitates the transfer of ubiquitin to lysine residues on the target protein, generating polyubiquitin chains. These polyubiquitinated proteins are then recognized and disassembled by the 26S proteasome complex [60].
In CHF patients, a cascade of pathophysiological changes alters levels of various hormones and inflammatory factors, which can upregulate atrophy-related genes such as Trim63 and Fbxo32. This upregulation enhances protein degradation while suppressing synthesis, ultimately leading to skeletal muscle atrophy. Ang-II, for instance, indirectly influences the UPS by generating an excess of ROS, which in turn activates the NF-κB signaling pathway [56]. Elevated levels of inflammatory cytokines like TNF-α, IL-1, and IL-6 can increase the transcription of atrogenes encoding muscle-specific E3 ligases, such as MuRF1 and MAFbx/atrogin-1, via NF-κB pathway, [59,61] and AMPK-Foxo pathway [62]. The suppression of Akt-mediated phosphorylation of Foxo transcription factor also contributes to the enhanced transcription of MuRF1 and MAFbx/atrogin-1 in cachexia [54,63]. Myostatin plays a dual role by promoting the transcription of Trim63/MuRF1 and Fbxo32 and proteolysis through the activin A receptor type IIB (ActRIIB) and the subsequent phosphorylation of SMAD2/3 in both skeletal and cardiac muscles [10]; it also inhibits muscle growth by preventing satellite cell proliferation and differentiation [64]. Furthermore, increased glucocorticoid receptor engagement also contributes to upregulation of Trim63 and Fbxo32, leading to intensified protein degradation [65].
AUTOPHAGY-LYSOSOME SYSTEM (ALS):
Concurrent with cachexia, the maintenance of skeletal muscle protein homeostasis involves cathepsin L-dependent degradation processes [66]. Within skeletal myocytes, the ALS and UPS are activated by signaling pathways such as AMPK, as well as Foxo3 and Foxo1. An accumulation of autophagy signals under catabolic conditions is associated with ensuing muscle atrophy [38,67].
APOPTOSIS:
Cardiac cachexia is associated not only with increased apoptosis in cardiac myocytes [68], but also, as Adams et al have identified [69], with heightened apoptosis in the skeletal muscles of over half of patients with congestive heart failure. This condition correlates with a reduction in exercise endurance. During this state, cells encounter intrinsic apoptotic stimuli, leading to mitochondrial damage and the release of cytochrome c mediated by the BCL-2 protein family. Concurrently, there is an upregulation of pro-apoptotic BAX (BCL2-Associated X) and a downregulation of anti-apoptotic BCL-2. These alterations trigger a chain reaction that promotes apoptosis through activation of caspase-9 and caspase-3 [69–71].
Structure and Functions of MuRF1
STRUCTURE:
E3 ubiquitin ligases are categorized into 3 types based on their structural domains: those with a RING-finger domain, those homologous to E6-associated protein C terminus (HECT), and those with an in-between RING (RING-in-Between-RING) domain that exhibit characteristics of both RING-finger and HECT-type E3s [73]. MuRF1, a RING-finger type E3 within the RING-B-box-Coiled-Coil (RBCC) family – also known as the Tripartite Motif (Trim) family [74] – serves as a connector, facilitating the transfer of ubiquitin from E2 enzymes to target proteins without direct ubiquitin binding [73].
Centner et al were the first to suggest MuRF1’s interaction with titin at the M-line and Z-line of muscle sarcomeres, regulating titin’s stability [75]. MuRF1 is predominantly found in striated muscles, including skeletal [18] and cardiac muscles, and it is also present in certain smooth muscles, potentially playing a role in postpartum uterine atrophy [76].
In skeletal muscles, MuRF1 selectively binds to and targets substrates such as myosin heavy chain (MyHC) [77,78], myosin light chain (MyLC) [79], myosin binding protein C (MyBP-C) [80], α-actin [81], and titin-cap protein (Titin-cap, or TCAP, or telethonin) [82] for ubiquitination and subsequent degradation. In cardiac myocytes, MuRF1’s localization includes the M-line and specific sequences within titin [83]. It targets MyHC [84], MyBP-C [85], and troponin I for ubiquitination [86].
FUNCTIONS:
The initial step in understanding MuRF1’s role in skeletal and cardiac muscle tissues is to manipulate the expression of its encoding gene, Trim63. This research is crucial for laying the groundwork for development of inhibitors targeting MuRF1. Studies on MuRF1-KO mice have demonstrated protection against muscle wasting across different atrophy models regardless of the muscle fiber type. Bodine et al found that MuRF1-KO mice retained 36% more muscle mass in the gastrocnemius after 14 days of denervation compared to wild-type mice [18]. In atrophy models induced by amino acid deprivation, Koyama et al observed enhanced protein synthesis in the quadriceps muscles of MuRF1-KO [87]. Labeit et al reported almost complete resistance to atrophy in the soleus muscles of MuRF1-KO mice after 10 days in a hindlimb suspension model [88]. Additionally, Baehr et al discovered that MuRF1-KO mice were resistant to glucocorticoid (dexamethasone)-induced muscle atrophy, maintaining muscle fiber cross-sectional area (CSA) over 2 weeks [21].
MuRF1 plays a multifaceted protective role in cardiac muscle tissue, distinct from its function in skeletal muscle. Its contributions to myocardial health are evident in 4 main areas: (1) Regulation of pathological myocardial remodeling: MuRF1-KO mice exhibit more pronounced cardiac hypertrophy post-Transverse Aortic Constriction (TAC) compared to wild-type mice, indicating that MuRF1 can mitigate compensatory myocardial changes during early heart failure and thus improve prognosis [89,90]. MuRF1 appears to suppress protein synthesis linked to hypertrophy, as evidenced by the exacerbation of left ventricular hypertrophy following pressure overload in MuRF1-KO mice [91]. Molecular pathways through which MuRF1 exerts this effect include downregulation of the CnA (Calcineurin A)-NFAT pathway, inhibition of the JNK/AP-1 signaling pathway, and interactions with serum response factor (SRF) that modulate hypertrophic gene expression [90,92–94]. (2) Association with hypertrophic cardiomyopathy: Chen et al sequenced the Trim63 gene in 302 hypertrophic cardiomyopathy patients and identified mutations that cause mislocalization of MuRF1 in cardiomyocytes, impair its ubiquitination activities, and lead to myocardial hypertrophy [84]. (3) Response to drug-induced myocardial atrophy: In MuRF1-KO mice, myocardial atrophy induced by drugs like dexmedetomidine (a2-adrenoceptor agonists), dexamethasone (glucocorticoids), and Adriamycin (antibiotic) was not observed to be beneficial [89,90,95]. (4) Protection against myocardial ischemia-reperfusion (I/R) injury: Overexpression of MuRF1 may protect against I/R damage by reducing cardiac myocyte apoptosis through the inhibition of the JNK signaling pathway and downstream AP-1 signaling [96].
Development of Related Inhibitors Targeting MuRF1
P013222:
Eddins et al were the pioneers in identifying the MuRF1 inhibitor P013222, first reported in 2011 [104]. Utilizing an E3 ligase screening platform, they assessed a diverse library of small molecules for MuRF1 activity inhibition and found that P013222 impeded MuRF1’s autoubiquitination in a dose-dependent manner. Furthermore, the inhibitor was shown to protect MyHC from degradation in dexamethasone-treated C2C12 myotubes, suggesting its ability to block MuRF1-dependent substrate ubiquitination. Despite these promising in vitro results, in vivo and clinical trial data for P013222 are not yet available.
ID#704946:
Bowen et al developed a small-molecule compound, ID#704946, which targets the MuRF1-Titin complex [105]. In myotube culture experiments, this compound exhibited minimal toxicity and effectively protected myotubes from atrophy induced by synthetic glucocorticoids such as dexamethasone. Animal trials involved C57/BL6 mice, which were divided into 3 groups to establish a cardiac cachexia model via weekly injections of 600 mg/kg monocrotaline (MCT), leading to right ventricular insufficiency due to pulmonary hypertension. The experimental group received the inhibitor starting 1 week before MCT administration. After 6 weeks, the mice were euthanized for skeletal muscle analysis. Tissue studies revealed that ID#704946 specifically inhibited the upregulation of MuRF1 expression induced by cardiac cachexia without affecting MAFbx/atrogin-1 expression. Furthermore, it impeded proteasome activity and the ubiquitination of contractile proteins, reducing actin depletion. In a rat model of cardiac cachexia, the compound significantly mitigated contractile dysfunction in the tibialis anterior (TA) and diaphragm muscles, although it did not positively affect weight loss or ventricular hypertrophy development.
In continued research with compound ID#709496, Bowen et al [106] addressed diaphragm myopathy and systolic dysfunction induced by CHF. They induced CHF in female C57/BL6 mice by ligating the anterior descending branch of the left coronary artery to stimulate myocardial infarction. The mice that subsequently developed diaphragm malfunction were divided into 2 groups: one receiving normal feed and the other receiving feed supplemented with the compound, in addition to a sham-operated group with normal feeding. By week 10, the experimental group exhibited signs of diaphragmatic disease, marked by a 21% decrease in cross-sectional area (CSA) and diminished maximum diaphragmatic contractility. Treatment with the compound significantly reversed the impairment in diaphragmatic contractility, although it did not have a notable effect on CSA.
The aforementioned studies not only underscored ID#704946’s effectiveness in preventing skeletal muscle wasting but also explored its mechanisms of action: (1) In the skeletal muscle of control mice, an increase in cardiac ankyrin repeat protein (CARP) expression was observed, a protein associated with the structural domain where MuRF1 acts on Titin and is present in both cardiac and skeletal muscle tissues [107]. Typically inactive, CARP overexpression is induced by pathogenic stimuli and is associated with muscular atrophy [108]. The experimental results suggest that ID#704946 can inhibit CARP overexpression, which is linked to MuRF1 activity in cardiac cachexia [105]. (2) Additionally, the studies proposed that ID#704946 maintains protein synthesis and reduces apoptosis in skeletal muscle cells, with an upregulation of ELF2B (E47-like factor 2) and downregulation of BAX being observed. However, further research is needed to verify the specificity of these effects in skeletal muscle [105]. (3) A subsequent study hypothesized that ID#704946 can affect mitochondrial metabolic balance [106]. Molecular analyses indicated that the activity and concentration of key mitochondrial enzymes – such as citrate synthase (CS), succinodehydrogenase (SUH), and oxidative respiratory chain complex I (Complex I) – were significantly reduced in CHF conditions. Treatment with ID#704946 improved these mitochondrial changes, suggesting that MuRF1 acts as a negative regulator of mitochondrial metabolic functions. While the study confirmed the beneficial impact on mitochondrial, the precise mechanisms by which ID#704946 alters mitochondrial metabolism were not extensively investigated. (4) Transcriptome analysis of C2C12 myotubes treated with ID#704946 revealed a decrease in MuRF1 expression by 4–10%, further supporting the compound’s potential role as a MuRF1 inhibitor [106].
In conclusion, further investigation is essential to clarify the precise mechanisms behind ID#704946’s therapeutic actions. This includes determining whether its efficacy stems from a suitable reduction in MuRF1 expression and activity, the disruption of interactions with target proteins, or broader effects such as those on mitochondrial metabolism, protein synthesis, and apoptosis. Additionally, the compound’s potential regulatory impact on MuRF2 and the possibility of functional redundancy should be validated through further studies. Clinical trials for ID#704946 have not been conducted, as there is insufficient clarity regarding its bioavailability, metabolic system toxicity, and impact on cardiac remodeling, and there is a lack of comprehensive data from animal models.
MYOMED-946 AND MYOMED-205:
Building on the success of compound ID#709496 in mitigating skeletal muscle wasting associated with cardiac cachexia [105] and diaphragm dysfunction in CHF [106], Adams et al synthesized 2 new compounds: MyoMed-946, which is structurally identical to ID#709496 but produced via a different synthesis method, and MyoMed-205, a chemically modified variant of ID#709496 designed to enhance serum stability [109]. These compounds were independently administered to mice with advanced melanoma to assess their impact on skeletal muscle function. Both MyoMed-946 and MyoMed-205 were reported to reduce weight loss in the TA, soleus, and extensor digitorum longus (EDL) muscles, indicating an improvement in muscle wasting conditions. Additionally, the mice showed protection against tumor-induced weight loss. Further analysis of EDL muscle tissue revealed that treatment with the inhibitors increased the expression of SMDT1, a calcium-regulatory subunit of the mitochondrial single transporter, potentially affecting mitochondrial apoptosis via the BAX pathway [109–111]. The compounds were also shown to prevent depletion of citrate synthase and maintain Complex I enzyme activity, suggesting that their beneficial effects on skeletal muscle wasting are due to a combination of mechanisms involving apoptosis regulation, mitochondrial energy metabolism, and MuRF1 inhibition.
In their subsequent research, Adams et al extended their investigation to the impact of MyoMed-205 on various skeletal muscle wasting disorders and cardiac function in HFpEF rats [32]. They divided 30 ZSF1 obese rats into 2 groups: one group received MyoMed-205 and the other served as a placebo group, with healthy mice acting as controls. After feeding the rats until 20 32 weeks to progressively induce the HFpEF model, the following conclusions were drawn from comprehensive tissue studies: (1) MyoMed-205 significantly reduced skeletal muscle atrophy to normal levels, particularly in the TA muscle, where it also increased muscle mass and CSA by 26%. The treatment led to decreased ubiquitination of total muscle proteins and a reduction in MuRF1 expression in skeletal muscle. (2) The compound mitigated mitochondrial stress effects from heart failure, as shown by proteomic analysis, which indicated differential expression of 8 proteins in the placebo group’s muscle tissue, of which 5 are related to mitochondrial metabolism and autophagy. (3) Treatment with MyoMed-205 resulted in an upregulation of protein synthesis for mitochondrial Complex I–V and significantly higher citrate synthase activity in the TA muscle, supporting the notion that MuRF1 affects mitochondrial oxidative phosphorylation, as demonstrated by a MuRF1-KO model. (4) MyoMed-205 caused greater improvement in skeletal muscle wasting compared to ID#704946, potentially attributable to its enhanced serum stability.
Recently, the research team led by Ribeiro has utilized MyoMed-205 in the therapeutic intervention of contractile impairments after unilateral phrenic nerve denervation, reaffirming the muscle-protective role of MuRF1 inhibitors concerning atrophy and muscle fiber cross-sectional dimensions. Expanding on this, they meticulously crafted experiments to assess the toxicity and dose-response profile of this MuRF1-targeting small molecule, with no adverse toxic effects noted within the dosage spectrum of 300 to 2000 mg/kg. Upon subjecting the diaphragm to denervation for 12 hours, a range of MyoMed-205 dosages (from 12.5 to 250 mg/kg body weight) was applied. Notably, the 50 mg/kg body weight dosage of MyoMed-205 demonstrated significant prevention of diaphragmatic contractile dysfunction, while higher dosages, such as 100 and 150 mg/kg body weight, provided partial amelioration of strength degradation. Conversely, the maximum dosage of 250 mg/kg body weight failed to manifest any protective impact on diaphragmatic strength. Western blotting analysis revealed that MyoMed-205, through the modulation of muscle atrophy-associated factors (HDAC4, FoxO1, and MuRF2) coupled with the activation of the Akt signaling cascade, confers a protective influence on the early-stage diaphragmatic dysfunction and atrophy [112].
Discussion
Effective treatment for cardiac cachexia with skeletal muscle wasting should encompass a multimodal approach, including pharmaceutical therapy, exercise training, and nutritional counseling. Recent advances in MuRF1 research have led to development of several small-molecule inhibitors targeted at this protein, such as ID#709496, MyoMed-946, and MyoMed-205, created by Adams’ team. These inhibitors have demonstrated potential in reducing skeletal muscle wasting in cachectic states.
Notably, In their investigation of the HFpEF rat model, Adams et al [32] examined the direct effects of MyoMed-205 on myocardial diastolic function. The compound decreased the severity of left ventricular sclerosis and improved cardiac diastolic function in the experimental rats, suggesting a potential reversal of myocardial alterations in HFpEF. Myocardial fibrosis, characterized by an excessive accumulation of extracellular matrix, plays a crucial role in myocardial remodeling in HFpEF. This process is regulated by an imbalance between matrix metalloproteinases (MMPs) and their tissue inhibitors, with MMP activation leading to reduced tensile strength of cardiac tissue and consequent diastolic dysfunction [113–116]. Treatment with MyoMed-205 resulted in reduced myocardial fibrosis in the experimental group compared to the placebo group, as indicated by lower MMP levels in protein blotting assays [32]. Furthermore, previous studies have linked low phosphorylation levels of Titin and an altered collagen-Titin ratio with the development of the HFpEF myocardium [117,118]. Adams et al found that in obese HFpEF mice, myocardial Titin phosphorylation levels and myocardial tissue collagen were low, although the experimental group did not show significant differences from the control group in myocardial phosphorylation levels [32]. They hypothesized that MyoMed-205 maintains myosin flexibility by promoting cGMP-PK (cyclic guanosine monophosphate-protein kinase G) phosphorylation modifications, a hypothesis that warrants further research. In summary, the study suggests that the beneficial effects of MyoMed-205, a small molecule targeting HFpEF, on cardiac diastolic function may be linked to reduced myocardial fibrosis and modifications in actin phosphorylation. The proposed mechanisms involve inhibition of MuRF1 expression, which reduces myogenic fibronectin degradation in the UPS pathway, balancing protein synthesis and degradation to prevent muscle wasting. Additionally, modulating actin and reducing fibrosis may benefit cardiac muscle during HFpEF.
The striated muscle groups in cardiac and skeletal muscle tissues are not the same, and MuRF1 performs distinct functions and has different domains of action in these 2 muscles, as previously described. Although MuRF1 plays a part in the atrophy of skeletal muscle, its function in cardiac muscle seems to be more complex. Further research into MuRF1’s function and mechanism in cardiac tissue is needed, particularly its influence on myocardial remodeling, ischemia-reperfusion injury, and myocardial hypertrophy [84,119,120].
Developing MuRF1-targeting inhibitors has several challenges: (1) There is a lack of comprehensive data from animal models and clinical trials, despite the demonstrated efficacy of these inhibitors in treating muscle wasting. (2) All inhibitors have not shown concentration-related negative effects, which could be due to the short duration of studies and their dissimilarity from typical clinical treatment courses. (3) The impact of MuRF1 suppression on the development of myocardial hypertrophy, a condition that worsens heart failure, is yet to be determined. Furthermore, MuRF1 plays a protective role against myocardial ischemia-reperfusion injury [96], raising concerns about potential exacerbation of reperfusion injury if MuRF1 expression is reduced. (4) Incomplete pharmacological, toxicological, and pharmacokinetic studies have hindered the progression and clinical application of MuRF1 inhibitors. (5) There are various subtypes within the MuRF protein family. In normal conditions, MuRF1 and MuRF2 co-knockout animals display cardiac and skeletal muscle hypertrophy [121], while MuRF3-KO mice do not show a distinct phenotype under ordinary conditions but have increased susceptibility to heart rupture after myocardial infarction. Co-knockout of MuRF1 and MuRF3 results in an abnormal accumulation of MyHC fragments in skeletal and cardiac muscle [122]. This suggests that MuRF1 and other family members share functions, and due to MuRF1’s complex transcriptional regulatory system, the development of inhibitors must be refined to enhance specificity towards MuRF1.
Despite the current absence of effective pharmacological treatments for patients with skeletal muscle wasting in cardiac cachexia, the identification of MuRF1-targeting inhibitors has opened up new therapeutic possibilities. While these inhibitors have limitations, their role in treating skeletal muscle wasting is promising, and using them as a novel target is currently a viable concept. Concurrently, establishing reliable diagnostic criteria for cardiac cachexia with skeletal muscle wasting is essential. A proactive approach in preventing skeletal muscle wasting in CHF patients, with a focus on MuRF1 as a key target, could potentially improve both the prognosis and the quality of life for these individuals. Traditional Chinese medicine has also explored this class of E3 ligases. Compounds such as ginsenoside Rg1 have shown potential to suppress MuRF1 expression in C2C12 cells [123], and formulations like the heated kidney and blood activation formula have improved cardiac function and exercise tolerance while reducing MAFbx/atrogin-1 and MuRF1 mRNA expression in heart failure models [124]. The E3 ligase encoded by the atrophy gene MAFbx/atrogin-1, which shares a similar transcriptional signaling pathway with MuRF1 [17,125–127], is another potential target against skeletal muscle wasting and myocardial injury. Investigating atrogenes like MuRF1/Trim63, MAFbx/atrogin-1/Fbxo32, and their homologs [128] histologically will not only elucidate their tissue action mechanisms but will also assist in developing new biomarkers for diagnosis, risk stratification, prognosis assessment, and monitoring treatment efficacy.
Figures
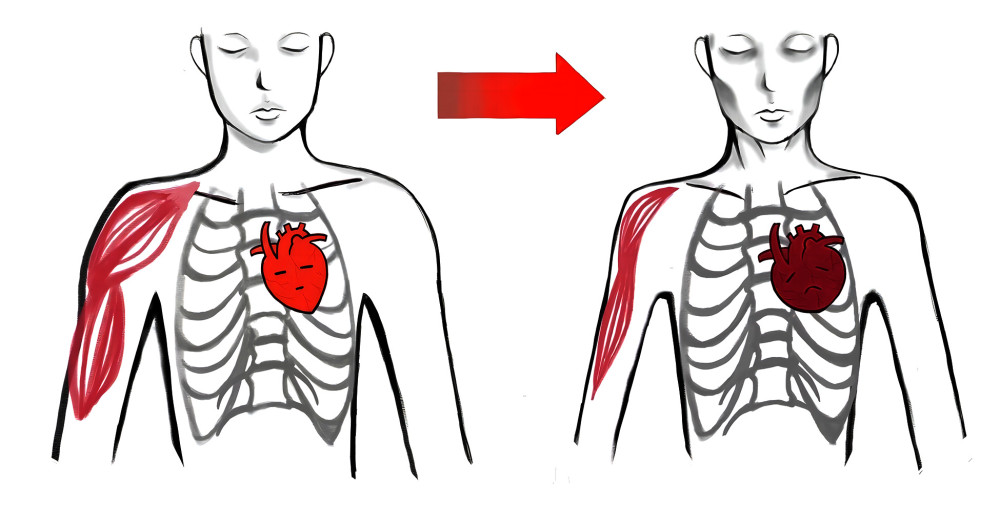 Figure 1. Cardiac cachexia represents an advanced stage of CHF characterized by systemic decline and metabolic dysfunction. This condition leads to profound nutrition depletion and a catabolic state, manifesting as anorexia, progressive weakness, emaciation, pronounced cheekbones, hollow eye sockets, a pallid complexion, and skeletal muscle atrophy. Produced using Procreate® developed by Savage Interactive Pty Ltd.(version5.2.6).
Figure 1. Cardiac cachexia represents an advanced stage of CHF characterized by systemic decline and metabolic dysfunction. This condition leads to profound nutrition depletion and a catabolic state, manifesting as anorexia, progressive weakness, emaciation, pronounced cheekbones, hollow eye sockets, a pallid complexion, and skeletal muscle atrophy. Produced using Procreate® developed by Savage Interactive Pty Ltd.(version5.2.6).  Figure 2. Wasting in CHF. CHF leads to a variety of pathological and physiological changes, causing continuous consumption of the body, resulting in skeletal muscle wasting, loss of bone mineral density, wasting to adipose tissue, and even leading to cachexia. Cachexia can also, in turn, promote more severe consumption. Produced using BioRender.com developed by BioRender Ltd.
Figure 2. Wasting in CHF. CHF leads to a variety of pathological and physiological changes, causing continuous consumption of the body, resulting in skeletal muscle wasting, loss of bone mineral density, wasting to adipose tissue, and even leading to cachexia. Cachexia can also, in turn, promote more severe consumption. Produced using BioRender.com developed by BioRender Ltd.  Figure 3. Signaling pathway with UPS-related muscle wasting in cardiac cachexiaIn cardiac cachexia, muscle wasting is driven by disruptions in the anabolic-catabolic balance. (1) During anabolic phases, IGF1 binds to the both the IGF-1 receptor (IGF1R) and the insulin receptor (IR), leading to the phosphorylation of insulin receptor substrate 1 (IRS1) and activation of the PI3K-Akt-mTOR signaling pathway, which promotes protein synthesis. However, this anabolic signaling is often impaired in CHF. (2) During catabolic phases, myostatin, pro-inflammatory cytokines such as TNFα, IL-1, and IL-6, Ang-II, and glucocorticoids bind to their respective receptors and initiate the transcription of atrophy-related genes Trim63 and Fbxo32 via the NF-κB, Foxo, and SMAD signaling pathways. This results in increased expression of E3 ubiquitin ligases, MuRF1, and MAFbx/atrogin-1, leading to protein degradation through UPS activation. Created with Microsoft® Excel® 2021MSO (version 2407 Build 16.0.17830.20166).
Figure 3. Signaling pathway with UPS-related muscle wasting in cardiac cachexiaIn cardiac cachexia, muscle wasting is driven by disruptions in the anabolic-catabolic balance. (1) During anabolic phases, IGF1 binds to the both the IGF-1 receptor (IGF1R) and the insulin receptor (IR), leading to the phosphorylation of insulin receptor substrate 1 (IRS1) and activation of the PI3K-Akt-mTOR signaling pathway, which promotes protein synthesis. However, this anabolic signaling is often impaired in CHF. (2) During catabolic phases, myostatin, pro-inflammatory cytokines such as TNFα, IL-1, and IL-6, Ang-II, and glucocorticoids bind to their respective receptors and initiate the transcription of atrophy-related genes Trim63 and Fbxo32 via the NF-κB, Foxo, and SMAD signaling pathways. This results in increased expression of E3 ubiquitin ligases, MuRF1, and MAFbx/atrogin-1, leading to protein degradation through UPS activation. Created with Microsoft® Excel® 2021MSO (version 2407 Build 16.0.17830.20166). 
References
1. Ponikowski P, Voors AA, Anker SDESC Scientific Document Group, 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC)Developed with the special contribution of the Heart Failure Association (HFA) of the ESC: Eur Heart J, 2016; 37; 2129-200
2. Doehner W, Anker SD, Cardiac cachexia in early literature: A review of research prior to Medline: Int J Cardiol, 2002; 85; 7-14
3. von Haehling S, Anker MS, Anker SD, Prevalence and clinical impact of cachexia in chronic illness in Europe, USA, and Japan: facts and numbers update 2016: J Cachexia Sarcopenia Muscle, 2016; 7; 507-9
4. Evans WJ, Morley JE, Argilés J, Cachexia: A new definition: Clin Nutr, 2008; 27; 793-99
5. Soto ME, Pérez-Torres I, Rubio-Ruiz ME, Interconnection between cardiac cachexia and heart failure-protective role of cardiac obesity: Cells, 2022; 11; 1039
6. Vest AR, Chan M, Deswal A, Nutrition, obesity, and cachexia in patients with heart failure: A consensus statement from the heart failure Society of America Scientific Statements Committee: J Card Fail, 2019; 25; 380-400
7. Anker SD, Ponikowski P, Varney S, Wasting as independent risk factor for mortality in chronic heart failure: Lancet Lond Engl, 1997; 349; 1050-53
8. Cohen S, Nathan JA, Goldberg AL, Muscle wasting in disease: Molecular mechanisms and promising therapies: Nat Rev Drug Discov, 2015; 14; 58-74
9. von Haehling S, The wasting continuum in heart failure: from sarcopenia to cachexia: Proc Nutr Soc, 2015; 74; 367-77
10. von Haehling S, Ebner N, Dos Santos MR, Muscle wasting and cachexia in heart failure: Mechanisms and therapies: Nat Rev Cardiol, 2017; 14; 323-41
11. Fülster S, Tacke M, Sandek A, Muscle wasting in patients with chronic heart failure: Results from the studies investigating co-morbidities aggravating heart failure (SICA-HF): Eur Heart J, 2013; 34; 512-19
12. Janssen I, Heymsfield SB, Ross R, Low relative skeletal muscle mass (sarcopenia) in older persons is associated with functional impairment and physical disability: J Am Geriatr Soc, 2002; 50; 889-96
13. Janssen I, Shepard DS, Katzmarzyk PT, The healthcare costs of sarcopenia in the United States: J Am Geriatr Soc, 2004; 52; 80-85
14. Taylor RS, Walker S, Ciani O, Exercise-based cardiac rehabilitation for chronic heart failure: the EXTRAMATCH II individual participant data meta-analysis: Health Technol Assess Winch Engl, 2019; 23; 1-98
15. Feng L, Li B, Xi Y, Aerobic exercise and resistance exercise alleviate skeletal muscle atrophy through IGF-1/IGF-1R-PI3K/Akt pathway in mice with myocardial infarction: Am J Physiol Cell Physiol, 2022; 322; C164-76
16. Bilodeau PA, Coyne ES, Wing SS, The ubiquitin proteasome system in atrophying skeletal muscle: Roles and regulation: Am J Physiol Cell Physiol, 2016; 311; C392-403
17. Bodine SC, Baehr LM, Skeletal muscle atrophy and the E3 ubiquitin ligases MuRF1 and MAFbx/atrogin-1: Am J Physiol Endocrinol Metab, 2014; 307; E469-84
18. Bodine SC, Latres E, Baumhueter S, Identification of ubiquitin ligases required for skeletal muscle atrophy: Science, 2001; 294; 1704-78
19. Lecker SH, Jagoe RT, Gilbert A, Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression: FASEB J, 2004; 18; 39-51
20. Allen DL, Cleary AS, Lindsay SF, Myostatin expression is increased by food deprivation in a muscle-specific manner and contributes to muscle atrophy during prolonged food deprivation in mice: J Appl Physiol (1985), 2010; 109; 692-701
21. Baehr LM, Furlow JD, Bodine SC, Muscle sparing in muscle RING finger 1 null mice: Response to synthetic glucocorticoids: J Physiol, 2011; 589; 4759-76
22. Wada S, Kato Y, Okutsu M, Translational suppression of atrophic regulators by microRNA-23a integrates resistance to skeletal muscle atrophy: J Biol Chem, 2011; 286; 38456-65
23. Sacheck JM, Hyatt J-PK, Raffaello A, Rapid disuse and denervation atrophy involve transcriptional changes similar to those of muscle wasting during systemic diseases: FASEB J, 2007; 21; 140-55
24. Zeman RJ, Zhao J, Zhang Y, Differential skeletal muscle gene expression after upper or lower motor neuron transection: Pflugers Arch, 2009; 458; 525-35
25. Hanson AM, Harrison BC, Young MH, Longitudinal characterization of functional, morphologic, and biochemical adaptations in mouse skeletal muscle with hindlimb suspension: Muscle Nerve, 2013; 48; 393-402
26. Chen Y-W, Gregory CM, Scarborough MT, Transcriptional pathways associated with skeletal muscle disuse atrophy in humans: Physiol Genomics, 2007; 31; 510-20
27. Gustafsson T, Osterlund T, Flanagan JN, Effects of 3 days unloading on molecular regulators of muscle size in humans: J Appl Physiol (1985), 2010; 109; 721-27
28. Hahn A, Kny M, Pablo-Tortola C, Serum amyloid A1 mediates myotube atrophy via Toll-like receptors: J Cachexia Sarcopenia Muscle, 2020; 11; 103-19
29. Chen L, Chen L, Wan L, Matrine improves skeletal muscle atrophy by inhibiting E3 ubiquitin ligases and activating the Akt/mTOR/FoxO3α signaling pathway in C2C12 myotubes and mice: Oncol Rep, 2019; 42; 479-94
30. Zhou X, Wang JL, Lu J, Reversal of cancer cachexia and muscle wasting by ActRIIB antagonism leads to prolonged survival: Cell, 2010; 142; 531-43
31. Usui S, Maejima Y, Pain J, Endogenous muscle atrophy F-box mediates pressure overload-induced cardiac hypertrophy through regulation of nuclear factor-kappaB: Circ Res, 2011; 109; 161-71
32. Adams V, Schauer A, Augstein A, Targeting MuRF1 by small molecules in a HFpEF rat model improves myocardial diastolic function and skeletal muscle contractility: J Cachexia Sarcopenia Muscle, 2022; 13(3); 1565-81
33. Brink M, Wellen J, Delafontaine P, Angiotensin II causes weight loss and decreases circulating insulin-like growth factor I in rats through a pressor-independent mechanism: J Clin Invest, 1996; 97; 2509-16
34. Whitehead NP, Yeung EW, Froehner SC, Skeletal muscle NADPH oxidase is increased and triggers stretch-induced damage in the mdx mouse: PLoS One, 2010; 5; e15354
35. Yoshida T, Semprun-Prieto L, Wainford RD, Angiotensin II reduces food intake by altering orexigenic neuropeptide expression in the mouse hypothalamus: Endocrinology, 2012; 153; 1411-20
36. Akamizu T, Kangawa K, Ghrelin for cachexia: J Cachexia Sarcopenia Muscle, 2010; 1; 169-76
37. Xin X, Ren A-J, Zheng X, Disturbance of circulating ghrelin and obestatin in chronic heart failure patients especially in those with cachexia: Peptides, 2009; 30; 2281-85
38. Yoshida T, Delafontaine P, Mechanisms of IGF-1-mediated regulation of skeletal muscle hypertrophy and atrophy: Cells, 2020; 9; E1970
39. Anker SD, Volterrani M, Pflaum CD, Acquired growth hormone resistance in patients with chronic heart failure: Implications for therapy with growth hormone: J Am Coll Cardiol, 2001; 38; 443-52
40. Krysztofiak H, Wleklik M, Migaj J, Cardiac cachexia: A well-known but challenging complication of heart failure: Clin Interv Aging, 2020; 15; 2041-51
41. Kistorp C, Faber J, Galatius S, Plasma adiponectin, body mass index, and mortality in patients with chronic heart failure: Circulation, 2005; 112; 1756-62
42. Lu W, Feng W, Lai J, Role of adipokines in sarcopenia: Chin Med J (Engl), 2023; 136; 1794-804
43. Doehner W, Pflaum CD, Rauchhaus M, Leptin, insulin sensitivity and growth hormone binding protein in chronic heart failure with and without cardiac cachexia: Eur J Endocrinol, 2001; 145; 727-35
44. Doehner W, Rauchhaus M, Godsland IF, Insulin resistance in moderate chronic heart failure is related to hyperleptinaemia, but not to norepinephrine or TNF-αlpha: Int J Cardiol, 2002; 83; 73-81
45. Vonhaehling S, Doehner W, Anker S, Nutrition, metabolism, and the complex pathophysiology of cachexia in chronic heart failure: Cardiovasc Res, 2007; 73; 298-309
46. Loncar G, Springer J, Anker M: J Cachexia Sarcopenia Muscle, 2016; 7; 246-60
47. Yamaji M, Tsutamoto T, Kawahara C, Serum cortisol as a useful predictor of cardiac events in patients with chronic heart failure: the impact of oxidative stress: Circ Heart Fail, 2009; 2; 608-15
48. Thanapholsart J, Khan E, Ismail TF, The complex pathophysiology of cardiac cachexia: A review of current pathophysiology and implications for clinical practice: Am J Med Sci, 2022; 365(1); 9-18
49. von Haehling S, Lainscak M, Springer J, Cardiac cachexia: A systematic overview: Pharmacol Ther, 2009; 121; 227-52
50. Anker SD, Ponikowski PP, Clark AL, Cytokines and neurohormones relating to body composition alterations in the wasting syndrome of chronic heart failure: Eur Heart J, 1999; 20; 683-93
51. Gaggin HK, Belcher AM, Gandhi PU, Serial echocardiographic characteristics, novel biomarkers and cachexia development in patients with stable chronic heart failure: J Cardiovasc Transl Res, 2016; 9; 429-31
52. Anker SD, Chua TP, Ponikowski P, Hormonal changes and catabolic/anabolic imbalance in chronic heart failure and their importance for cardiac cachexia: Circulation, 1997; 96; 526-34
53. Anker SD, von Haehling S, Inflammatory mediators in chronic heart failure: An overview: Heart Br Card Soc, 2004; 90; 464-70
54. Franch HA, Price SR, Molecular signaling pathways regulating muscle proteolysis during atrophy: Curr Opin Clin Nutr Metab Care, 2005; 8; 271-75
55. Vander Haar E, Lee S-I, Bandhakavi S, Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40: Nat Cell Biol, 2007; 9; 316-23
56. Sukhanov S, Semprun-Prieto L, Yoshida T, Angiotensin II, oxidative stress and skeletal muscle wasting: Am J Med Sci, 2011; 342; 143-47
57. Breitbart A, Auger-Messier M, Molkentin JD, Myostatin from the heart: Local and systemic actions in cardiac failure and muscle wasting: Am J Physiol Heart Circ Physiol, 2011; 300; H1973-82
58. Morissette MR, Cook SA, Buranasombati C, Myostatin inhibits IGF-I-induced myotube hypertrophy through Akt: Am J Physiol Cell Physiol, 2009; 297; C1124-32
59. Khalil R, Ubiquitin-proteasome pathway and muscle atrophy: Adv Exp Med Biol, 2018; 1088; 235-48
60. Willis MS, Townley-Tilson WHD, Kang EY, Sent to destroy: The ubiquitin proteasome system regulates cell signaling and protein quality control in cardiovascular development and disease: Circ Res, 2010; 106; 463-78
61. Yoshida T, Tabony AM, Galvez S, Molecular mechanisms and signaling pathways of angiotensin II-induced muscle wasting: Potential therapeutic targets for cardiac cachexia: Int J Biochem Cell Biol, 2013; 45; 2322-32
62. Webster JM, Kempen LJAP, Hardy RS, Inflammation and skeletal muscle wasting during cachexia: Front Physiol, 2020; 11; 597675
63. Glass DJ, Signaling pathways perturbing muscle mass: Curr Opin Clin Nutr Metab Care, 2010; 13; 225-29
64. Cohen S, Nathan JA, Goldberg AL, Muscle wasting in disease: Molecular mechanisms and promising therapies: Nat Rev Drug Discov, 2015; 14; 58-74
65. Schakman O, Kalista S, Barbé C, Glucocorticoid-induced skeletal muscle atrophy: Int J Biochem Cell Biol, 2013; 45; 2163-72
66. Deval C, Mordier S, Obled C, Identification of cathepsin L as a differentially expressed message associated with skeletal muscle wasting: Biochem J, 2001; 360; 143-50
67. Sanchez AMJ, Csibi A, Raibon A, AMPK promotes skeletal muscle autophagy through activation of forkhead FoxO3a and interaction with Ulk1: J Cell Biochem, 2012; 113; 695-710
68. Narula J, Haider N, Virmani R, Apoptosis in myocytes in end-stage heart failure: N Engl J Med, 1996; 335; 1182-89
69. Adams V, Jiang H, Yu J, Apoptosis in skeletal myocytes of patients with chronic heart failure is associated with exercise intolerance: J Am Coll Cardiol, 1999; 33; 959-65
70. Knezevic T, Myers VD, Gordon J, BAG3: A new player in the heart failure paradigm: Heart Fail Rev, 2015; 20; 423-34
71. Vescovo G, Dalla Libera L, Skeletal muscle apoptosis in experimental heart failure: The only link between inflammation and skeletal muscle wastage?: Curr Opin Clin Nutr Metab Care, 2006; 9; 416-22
72. Bodine SC, Baehr LM, Skeletal muscle atrophy and the E3 ubiquitin ligases MuRF1 and MAFbx/atrogin-1: Am J Physiol-Endocrinol Metab, 2014; 307; E469-84
73. Peris-Moreno D, Cussonneau L, Combaret L, Ubiquitin ligases at the heart of skeletal muscle atrophy control: Molecules Basel Switz, 2021; 26; E407
74. Mrosek M, Meier S, Ucurum-Fotiadis Z, Structural analysis of B-Box 2 from MuRF1: Identification of a novel self-association pattern in a RING-like fold: Biochemistry, 2008; 47; 10722-30
75. Centner T, Yano J, Kimura E, Identification of muscle specific ring finger proteins as potential regulators of the titin kinase domain 11: J Mol Biol, 2001; 306; 717-26
76. Bdolah Y, Segal A, Tanksale P, Atrophy-related ubiquitin ligases atrogin-1 and MuRF-1 are associated with uterine smooth muscle involution in the postpartum period: Am J Physiol Regul Integr Comp Physiol, 2007; 292; R971-76
77. Clarke BA, Drujan D, Willis MS, The E3 ligase MuRF1 degrades myosin heavy chain protein in dexamethasone-treated skeletal muscle: Cell Metab, 2007; 6; 376-85
78. Fielitz J, Kim MS, Shelton JM, Myosin accumulation and striated muscle myopathy result from the loss of muscle RING finger 1 and 3: J Clin Invest, 2007; 117; 2486-95
79. Witt SH, Granzier H, Witt CC, MURF-1 and MURF-2 target a specific subset of myofibrillar proteins redundantly: Towards understanding MURF-dependent muscle ubiquitination: J Mol Biol, 2005; 350; 713-22
80. Cohen S, Brault JJ, Gygi SP, Glass DJ, During muscle atrophy, thick, but not thin, filament components are degraded by MuRF1-dependent ubiquitylation: J Cell Biol, 2009; 185; 1083-95
81. Polge C, Heng A-E, Jarzaguet M, Muscle actin is polyubiquitinylated in vitro and in vivo and targeted for breakdown by the E3 ligase MuRF1: FASEB J, 2011; 25; 3790-802
82. Polge C, Cabantous S, Deval C, A muscle-specific MuRF1-E2 network requires stabilization of MuRF1-E2 complexes by telethonin, a newly identified substrate: J Cachexia Sarcopenia Muscle, 2018; 9; 129-45
83. McElhinny AS, Kakinuma K, Sorimachi H, Muscle-specific RING finger-1 interacts with titin to regulate sarcomeric M-line and thick filament structure and may have nuclear functions via its interaction with glucocorticoid modulatory element binding protein-1: J Cell Biol, 2002; 157; 125-36
84. Chen SN, Czernuszewicz G, Tan Y, Human molecular genetic and functional studies identify TRIM63, encoding Muscle RING Finger Protein 1, as a novel gene for human hypertrophic cardiomyopathy: Circ Res, 2012; 111; 907-19
85. Mearini G, Gedicke C, Schlossarek S, Atrogin-1 and MuRF1 regulate cardiac MyBP-C levels via different mechanisms: Cardiovasc Res, 2010; 85; 357-66
86. Kedar V, McDonough H, Arya R, Muscle-specific RING finger 1 is a bona fide ubiquitin ligase that degrades cardiac troponin I: Proc Natl Acad Sci USA, 2004; 101; 18135-40
87. Koyama S, Hata S, Witt CC, Muscle RING-finger protein-1 (MuRF1) as a connector of muscle energy metabolism and protein synthesis: J Mol Biol, 2008; 376; 1224-36
88. Labeit S, Kohl CH, Witt CC, Modulation of muscle atrophy, fatigue and MLC phosphorylation by MuRF1 as indicated by hindlimb suspension studies on MuRF1-KO mice: J Biomed Biotechnol, 2010; 2010; 693741
89. Willis MS, Rojas M, Li L, Muscle ring finger 1 mediates cardiac atrophy in vivo: Am J Physiol Heart Circ Physiol, 2009; 296; H997-1006
90. Willis MS, Ike C, Li L, Muscle ring finger 1, but not muscle ring finger 2, regulates cardiac hypertrophy in vivo: Circ Res, 2007; 100; 456-59
91. Willis MS, Schisler JC, Li L, Cardiac muscle ring finger-1 increases susceptibility to heart failure in vivo: Circ Res, 2009; 105; 80-88
92. Molkentin JD, Calcineurin-NFAT signaling regulates the cardiac hypertrophic response in coordination with the MAPKs: Cardiovasc Res, 2004; 63; 467-75
93. Maejima Y, Usui S, Zhai P, Muscle-specific RING finger 1 negatively regulates pathological cardiac hypertrophy through downregulation of calcineurin A: Circ Heart Fail, 2014; 7; 479-90
94. Wadosky KM, Rodríguez JE, Hite RL, Muscle RING finger-1 attenuates IGF-I-dependent cardiomyocyte hypertrophy by inhibiting JNK signaling: Am J Physiol Endocrinol Metab, 2014; 306; E723-39
95. Willis MS, Parry TL, Brown DI, Doxorubicin exposure causes subacute cardiac atrophy dependent on the striated muscle-specific ubiquitin ligase MuRF1: Circ Heart Fail, 2019; 12; e005234
96. Li H-H, Du J, Fan Y-N, The ubiquitin ligase MuRF1 protects against cardiac ischemia/reperfusion injury by its proteasome-dependent degradation of phospho-c-Jun: Am J Pathol, 2011; 178; 1043-58
97. Caron AZ, Haroun S, Leblanc E, The proteasome inhibitor MG132 reduces immobilization-induced skeletal muscle atrophy in mice: BMC Musculoskelet Disord, 2011; 12; 185
98. Kline WO, Panaro FJ, Yang H, Rapamycin inhibits the growth and muscle-sparing effects of clenbuterol: J Appl Physiol, 1985; 102; 740-47
99. Frost RA, Nystrom GJ, Jefferson LS, Hormone, cytokine, and nutritional regulation of sepsis-induced increases in atrogin-1 and MuRF1 in skeletal muscle: Am J Physiol Endocrinol Metab, 2007; 292; E501-12
100. Wang H, Lai Y-J, Chan Y-L, Epigallocatechin-3-gallate effectively attenuates skeletal muscle atrophy caused by cancer cachexia: Cancer Lett, 2011; 305; 40-49
101. Yakabe M, Ogawa S, Ota H, Inhibition of interleukin-6 decreases atrogene expression and ameliorates tail suspension-induced skeletal muscle atrophy: PLoS One, 2018; 13; e0191318
102. Belova SP, Mochalova EP, Kostrominova TY, P38α-MAPK signaling inhibition attenuates soleus atrophy during early stages of muscle unloading: Int J Mol Sci, 2020; 21; 2756
103. Gómez-SanMiguel AB, Gomez-Moreira C, Nieto-Bona MP, Formoterol decreases muscle wasting as well as inflammation in the rat model of rheumatoid arthritis: Am J Physiol Endocrinol Metab, 2016; 310; E925-37
104. Eddins MJ, Marblestone JG, Suresh Kumar KG, Targeting the ubiquitin E3 ligase MuRF1 to inhibit muscle atrophy: Cell Biochem Biophys, 2011; 60; 113-18
105. Bowen TS, Adams V, Werner S, Small-molecule inhibition of MuRF1 attenuates skeletal muscle atrophy and dysfunction in cardiac cachexia: J Cachexia Sarcopenia Muscle, 2017; 8; 939-53
106. Adams V, Bowen TS, Werner S, Small-molecule-mediated chemical knock-down of MuRF1/MuRF2 and attenuation of diaphragm dysfunction in chronic heart failure: J Cachexia Sarcopenia Muscle, 2019; 10(5); 1102-15
107. Stevens M, Franke B, Skorupka KA, Exploration of the TRIM fold of MuRF1 using EPR reveals a canonical antiparallel structure and extended COS-Box: J Mol Biol, 2019; 431; 2900-9
108. Laure L, Suel L, Roudaut C, Cardiac ankyrin repeat protein is a marker of skeletal muscle pathological remodelling: FEBS J, 2009; 276; 669-84
109. Adams V, Gußen V, Zozulya S, Small-molecule chemical knockdown of MuRF1 in melanoma bearing mice attenuates tumor cachexia associated myopathy: Cells, 2020; 9; E2272
110. Gottschalk B, Klec C, Leitinger G, MICU1 controls cristae junction and spatially anchors mitochondrial Ca2+ uniporter complex: Nat Commun, 2019; 10; 3732
111. Xie K-F, Guo D-D, Luo X-J, SMDT1-driven change in mitochondrial dynamics mediate cell apoptosis in PDAC: Biochem Biophys Res Commun, 2019; 511; 323-29
112. Ribeiro F, Alves PKN, Bechara LRG, Small-molecule inhibition of MuRF1 prevents early disuse-induced diaphragmatic dysfunction and atrophy: Int J Mol Sci, 2023; 24; 3637
113. de Boer RA, De Keulenaer G, Bauersachs J, Towards better definition, quantification and treatment of fibrosis in heart failure. A scientific roadmap by the Committee of Translational Research of the Heart Failure Association (HFA) of the European Society of Cardiology: Eur J Heart Fail, 2019; 21; 272-85
114. Maruyama K, Imanaka-Yoshida K, The pathogenesis of cardiac fibrosis: A review of recent progress: Int J Mol Sci, 2022; 23; 2617
115. Li L, Zhao Q, Kong W, Extracellular matrix remodeling and cardiac fibrosis: Matrix Biol J Int Soc Matrix Biol, 2018; 68–69; 490-506
116. Mujumdar VS, Smiley LM, Tyagi SC, Activation of matrix metalloproteinase dilates and decreases cardiac tensile strength: Int J Cardiol, 2001; 79; 277-86
117. Zile MR, Baicu CF, Ikonomidis JS, Myocardial stiffness in patients with heart failure and a preserved ejection fraction: contributions of collagen and titin: Circulation, 2015; 131; 1247-59
118. Hamdani N, Franssen C, Lourenço A, Myocardial titin hypophosphorylation importantly contributes to heart failure with preserved ejection fraction in a rat metabolic risk model: Circ Heart Fail, 2013; 6; 1239-49
119. Conraads VM, Vrints CJ, Rodrigus IE, Depressed expression of MuRF1 and MAFbx in areas remote of recent myocardial infarction: A mechanism contributing to myocardial remodeling?: Basic Res Cardiol, 2010; 105; 219-26
120. Adams V, Linke A, Wisloff U, Myocardial expression of Murf-1 and MAFbx after induction of chronic heart failure: Effect on myocardial contractility: Cardiovasc Res, 2007; 73; 120-29
121. Witt CC, Witt SH, Lerche S, Cooperative control of striated muscle mass and metabolism by MuRF1 and MuRF2: EMBO J, 2008; 27; 350-60
122. Fielitz J, Kim M-S, Shelton JM, Myosin accumulation and striated muscle myopathy result from the loss of muscle RING finger 1 and 3: J Clin Invest, 2007; 117; 2486-95
123. Li F-Y, Jia S-N, Ma S, Research on Ginsenoside Rg1 inhibits MuRF-1/Atrogin-1 expression in C2C12 cells: Journal of Changchun Normal University(Natural Science), 2017; 36; 65-70
124. Peng J, Peng S, Effects of warming kidney and activating blood formula on exercise tolerance and expression of Atrogin1/MAFbx and MuRF1 mRNA in heart failure rats: Lishizhen Medicine and Materia Medica Research, 2019; 30; 70-73
125. Lee D, Goldberg A, Atrogin1/MAFbx: What atrophy, hypertrophy, and cardiac failure have in common: Circ Res, 2011; 109; 123-26
126. Usui S, Maejima Y, Pain J, Endogenous muscle atrophy F-box mediates pressure overload-induced cardiac hypertrophy through regulation of nuclear factor-kappaB: Circ Res, 2011; 109; 161-71
127. Baskin KK, Rodriguez MR, MAFbx/Atrogin-1 is required for atrophic remodeling of the unloaded heart: J Mol Cell Cardiol, 2014; 72; 168-76
128. Banerjee R, He J, Spaniel C, Non-targeted metabolomics analysis of cardiac Muscle Ring Finger-1 (MuRF1), MuRF2, and MuRF3 in vivo reveals novel and redundant metabolic changes: Metabolomics, 2015; 11(2); 312-22
Figures
Figure 1. Cardiac cachexia represents an advanced stage of CHF characterized by systemic decline and metabolic dysfunction. This condition leads to profound nutrition depletion and a catabolic state, manifesting as anorexia, progressive weakness, emaciation, pronounced cheekbones, hollow eye sockets, a pallid complexion, and skeletal muscle atrophy. Produced using Procreate® developed by Savage Interactive Pty Ltd.(version5.2.6).Figure 2. Wasting in CHF. CHF leads to a variety of pathological and physiological changes, causing continuous consumption of the body, resulting in skeletal muscle wasting, loss of bone mineral density, wasting to adipose tissue, and even leading to cachexia. Cachexia can also, in turn, promote more severe consumption. Produced using BioRender.com developed by BioRender Ltd.Figure 3. Signaling pathway with UPS-related muscle wasting in cardiac cachexiaIn cardiac cachexia, muscle wasting is driven by disruptions in the anabolic-catabolic balance. (1) During anabolic phases, IGF1 binds to the both the IGF-1 receptor (IGF1R) and the insulin receptor (IR), leading to the phosphorylation of insulin receptor substrate 1 (IRS1) and activation of the PI3K-Akt-mTOR signaling pathway, which promotes protein synthesis. However, this anabolic signaling is often impaired in CHF. (2) During catabolic phases, myostatin, pro-inflammatory cytokines such as TNFα, IL-1, and IL-6, Ang-II, and glucocorticoids bind to their respective receptors and initiate the transcription of atrophy-related genes Trim63 and Fbxo32 via the NF-κB, Foxo, and SMAD signaling pathways. This results in increased expression of E3 ubiquitin ligases, MuRF1, and MAFbx/atrogin-1, leading to protein degradation through UPS activation. Created with Microsoft® Excel® 2021MSO (version 2407 Build 16.0.17830.20166). In Press
Clinical Research
Effects of Single-Bout Endurance Exercise Intensity on Peripheral Neurotrophic Factors in Patients With Isc...Med Sci Monit In Press; DOI: 10.12659/MSM.952089
Review article
Anisodus tanguticus in Cancer Research: A Review of Traditional Use, Phytochemistry, Extraction Methods, an...Med Sci Monit In Press; DOI: 10.12659/MSM.952999
Clinical Research
Nasal Mucociliary Clearance and Its Relationship With Disease Severity in Patients With Multiple SclerosisMed Sci Monit In Press; DOI: 10.12659/MSM.952850
Clinical Research
Modified Thoracoabdominal Nerves Block Through the Perichondrial Approach vs Subcostal Transversus Abdomini...Med Sci Monit In Press; DOI: 10.12659/MSM.953976
Most Viewed Current Articles
17 Jan 2024 : Review article 14,176,570
Vaccination Guidelines for Pregnant Women: Addressing COVID-19 and the Omicron VariantDOI :10.12659/MSM.942799
Med Sci Monit 2024; 30:e942799
13 Nov 2021 : Clinical Research 3,762,188
Acceptance of COVID-19 Vaccination and Its Associated Factors Among Cancer Patients Attending the Oncology ...DOI :10.12659/MSM.932788
Med Sci Monit 2021; 27:e932788
14 Dec 2022 : Clinical Research 2,466,310
Prevalence and Variability of Allergen-Specific Immunoglobulin E in Patients with Elevated Tryptase LevelsDOI :10.12659/MSM.937990
Med Sci Monit 2022; 28:e937990
16 May 2023 : Clinical Research 708,927
Electrophysiological Testing for an Auditory Processing Disorder and Reading Performance in 54 School Stude...DOI :10.12659/MSM.940387
Med Sci Monit 2023; 29:e940387