16 July 2020: Animal Study
Alisma Shugan Decoction (ASD) Ameliorates Hepatotoxicity and Associated Liver Dysfunction by Inhibiting Oxidative Stress and p65/Nrf2/JunD Signaling Dysregulation
Yunfeng Sun12AEG, Honghua Pan12CD, Shenghui Shen23ABCF, Zhongni Xia12BC, Zhongmin Yu2ABCD, ChengLe Li12F, Pingping Sun12DEG, Chuanwei Xin12DG*DOI: 10.12659/MSM.921738
Med Sci Monit 2020; 26:e921738






Abstract
BACKGROUND: Liver fibrosis, defined as the aberrant accumulation of extracellular matrix (ECM) proteins such as collagen in the liver, is a common feature of chronic liver disease, and often culminates in portal hypertension, liver cirrhosis, and hepatic failure. Though therapeutically manageable, fibrosis is not always successfully treated by conventional antifibrotic agents. While the traditional Chinese medicine (TCM) Alisma Shugan Decoction (ASD) has several health benefits, including anti-inflammation, anti-oxidation, and limitation of cardiovascular and respiratory disorders, it remains unclear if it has any hepato-protective potential.
MATERIAL AND METHODS: The present study examined the therapeutic effect of ASD in thioacetamide (TAA)-induced liver injury and fibrosis rat models.
RESULTS: We demonstrated that 50 mg/kg ASD significantly reversed TAA-induced elevation of alanine or aspartate transaminase levels, elicited no dyscrasia, and conferred a 40% (p<0.01) or 20% (p<0.05) survival advantage, compared to rats treated with TAA or TAA+ASD, respectively. Treatment with ASD reversed TAA-induced liver injury and fibrogenesis via repression of α-SMA protein and reduction of the collagen area and fibrosis score. Concurrently, ASD markedly suppressed the mRNA expression of fibrogenic procollagen, ICAM-1, MMP2, MMP9, and MMP13, and production of TIMP-1, ICAM-1, CXCL7, or CD62L cytokine in rat liver injury models. Interestingly, ASD-elicited reduction of liver injury and fibrogenesis was mediated by dysregulated p65/NrF-2/JunD signaling, with a resultant 3.18-fold (p<0.05) increase in GSH/GSSH ratio, and a 3.61-fold (p<0.01) or 1.51-fold (p<0.01) reduction in the 4-hydroxynonenal and malondialdehyde (MDA) levels, respectively, indicating reduced oxidative stress in the ASD-treated rats, and suggesting an hepato-protective role for ASD.
CONCLUSIONS: In conclusion, the present study provides supplementary evidence of the therapeutic benefit of ASD as an efficient treatment option in cases of liver injury and fibrosis. Further large-cohort validation of these findings is warranted.
Keywords: Adenoma, Liver Cell, Fibrosis, herbal medicine, Phytotherapy, Alisma, Atractylodes, hepatic stellate cells, Liver Cirrhosis, Liver Diseases, Oxidation-Reduction
Background
Chronic liver diseases are often associated with excessive deposition of extracellular matrix (ECM) proteins such as collagen and α-SMA, disintegration of hepatic parenchyma, distortion of liver architecture, and fibrous scarification, collectively referred to as liver fibrosis [1], especially as severe liver fibrosis results in end-stage liver disorders, including cirrhosis, liver failure, and portal hypertension, necessitating liver transplantation [1]. Mechanistically, subsequent to any acute liver injury, such as in viral hepatitis or drug toxicity, regeneration of the cellular components of the liver parenchyma is initiated to replace apoptotic or necrotic cells, with concomitant activation of inflammatory response and deposition of ECM. In cases where liver injury persists and liver regeneration is insufficient, liver cells are then replaced with excessive ECM, including fibrillar collagen [1–4]. Liver fibrosis remains a global clinical challenge due to lack of effective therapies, except for liver transplantation, which continues to be the only treatment available for patients with advanced fibrosis [5]. Increasing evidence suggests that hepatic fibrosis can be reversed by removal or repression of the underlying cause, thus necessitating the discovery of novel antifibrotic therapeutic approaches that can effectively and efficiently prevent, halt, or reverse fibrosis [5,6].
It is currently thought that the chronic induction of wound-healing reaction, oxidative stress (OS), and impaired epithelial-mesenchymal interaction in the pathophysiology of liver fibrosis are associated with the generation of reactive cholangiocytes and peribiliary fibrosis [7]. OS reflects the imbalance between produced reactive oxygen species (ROS) and the efficiency of antioxidant scavengers. Aberrant generation ROS from many innate oxidative enzymes disrupts liver-specific cell physiology and homeostasis, while probably playing a role in the pathogenesis of liver fibrosis; in fact, ROS production has been shown to play a critical role in liver injury and fibrogenesis through induction of apoptosis and/or necrosis of hepatocytes, amplification of associated inflammatory response, enhanced production of fibrogenic mediators by Kupffer-Browicz stellate macrophages or circulating inflammatory cells, and activation of hepatic stellate cells [8,9]. As OS continues to be associated with the initiation and progression of fibrosis, and OS-related molecules have been suggested to be mediators of bio-cellular events involved in liver fibrosis, the therapeutic promise of targeting OS-related signaling and associated molecular pathways in the management of liver injury and fibrosis continues to be explored [9] and is the subject of the present study.
This present study evaluated the effect of ASD on the liver function and phenotype of rats bearing TAA-induced hepatic injury and fibrosis, and we also investigated the probable crosstalk between OS and liver inflammatory cytokines, and the relationship with liver cell death. For the first time, to the best of our knowledge, we show that ASD downregulates pathological liver inflammatory response and deactivates oxidation stress to inhibit development of TAA-induced hepatic fibrosis in a rat model. Our results provide some mechanistic insights into the therapeutic effects of ASD on chemically-induced stress-related hepatic fibrosis, and highlight the protective effect of ASD in the fibrotic reaction to chronic hepatic injury.
Material and Methods
PREPARATION OF WATER EXTRACT FROM ALISMA SHUGAN DECOCTION (ASD):
Alisma Shugan Decoction (ASD) is a classical traditional Chinese formula that was first prescribed in the Eastern Han Dynasty, which consists of a combination of 2 herbs:
:
We obtained 2–3-month-old Wistar albino male rats (n=30) weighing 195–230 g from Shanghai SLAC Laboratory Animal Co. (Shanghai, China). Mice were acclimatized at 23±3.5°C with 54±6% humidity and a controlled 12: 12 h light-dark cycle. The rats were housed in a comfortable environment and were allowed free access to water and laboratory rodent chow. We randomly selected 10 rats and assigned to the untreated control group, while the remaining 20 rats were injected with 250 mg/kg thioacetamide (TAA) biweekly for 6 weeks to induce liver injury and fibrosis, and then they were randomly allocated into 1 of 2 groups: the TAA alone group (n=10) or the TAA+ASD group (n=10). ASD was administered intraperitoneally at 50 mg/kg/day, every week, starting on day 15 after TAA induction. Tumor growth, rat body weight, and survival were monitored throughout the experiment. Weights of harvested livers were assessed at the end of the experiment. The rats were anesthetized with ketamine/xylazine cocktail and sacrificed by CO2 euthanasia at the end of the study to allow harvesting and weighing of organs of interest. Livers were harvested 24 h after the last TAA injection on day 45 and weighed. The study protocol was carried out in strict accordance with the recommendations of the Laboratory Animals Committee and was specially approved by the Ethics Committee (LAC-2019-0601).
ANALYSIS OF BIOCHEMICAL PARAMETERS (ALT/AST, GSH/GSSG, MDA, 4-HNE):
The serum ALT or AST were estimated using an alanine transaminase activity assay kit (colorimetric/fluorometric) (ab105134) or aspartate aminotransferase activity assay kit (colorimetric) (ab105135), respectively, purchased from Abcam (Abcam, Cambridge, UK), strictly following the manufacturer’s instructions. For glutathione (GSH)/glutathione disulfide (GSSG) ratio, we used the GSH/GSSG ratio detection assay kit (Fluorometric – Green) (ab138881; Abcam, Cambridge, UK) according to the manufacturer’s instructions. Liver malondialdehyde (MDA) or 4-hydroxynonenal Colorimetric/fluorometric Lipid peroxidation (MDA) assay kits (ab118970) or colorimetric Lipid peroxidation (4-HNE) assay kits (ab238538) were used following the manufacturer’s instructions.
HISTOPATHOLOGICAL ANALYSIS:
After fixation of tissue samples in 4% formalin and paraffin embedding according to standard protocols, hematoxylin and eosin (H&E) staining of 5-μm-thick paraffin-embedded tissue blocks was performed for comparative histological analysis. Liver sections were stained with the Picro Sirius red stain kit (connective tissue stain) (ab150681; Abcam plc., Cambridge, UK) for histological visualization of collagen fibers and hepatic fibrosis following the manufacturer’s instructions. For α-SMA staining, anti-alpha smooth muscle actin antibody purchased from Abcam (ab15734; Abcam plc., Cambridge, UK) was used. Sections were visualized, and representative images taken under a standard light microscope (Leica Microsystems, [SEA] Pte, Singapore). Area percentage of collagen fibers per surface area in liver tissue was morphometrically estimated using Leica Qwin v2.0 image analysis software (Leica Imaging Systems, Cambridge, UK) in 5 high-power microscopic fields (hpf) per tissue section from rats from all treatment groups. The extent of liver fibrosis was determined based on the Ishak fibrosis score [11] by 2 pathologists in a blinded fashion.
WESTERN BLOT ANALYSIS:
For Western blot analysis, following the homogenization of liver tissue samples in 1 mL RIPA buffer containing protease and phosphatase inhibitor cocktails at 4°C, the homogenate was incubated on ice for 30 min, centrifuged at 4°C and 13 000 g for 30 min, and supernatant was obtained and stored in aliquots at –80°C until use. Thereafter, the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific, Waltham, MA, USA) was used to determine protein concentrations following the manufacturer’s instructions, and equal amounts (20 μg) of protein were separated by 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and blots transferred to polyvinylidene difluoride (PVDF) membranes (Millipore, Bedford, MA). The membranes were blocked at 37°C with 5% non-fat milk in Tris-buffered saline containing 0.05% Tween-20 (TBST) for 30 min, and incubated with primary antibodies against α-SMA (ab5694; Abcam, Cambridge, UK), Nrf-2 (ab62352; Abcam, Cambridge, UK), NF-κB p65 (ab16502; Abcam, Cambridge, UK), JunD (ab28837; Abcam, Cambridge, UK), PCNA (10205-2-AP; PROTEINTECH), or α-tubulin (66031-1-Ig; PROTEINTECH) overnight at 4°C (Supplementary Table 1). After carefully washing 3 times with TBST, the membranes were incubated for 1 h with anti-mouse or anti-rabbit secondary HRP-conjugated antibody at room temperature, and protein signals visualized using an enhanced chemiluminescence (ECL) detection kit (GE Healthcare, RPN2108, Sigma, Merck KGaA, Darmstadt, Germany).
MEASUREMENT OF SERUM TIMP-1, ICAM-1, CXCL7, OR CD62L CYTOKINE LEVELS:
The serum levels of tissue inhibitor of metalloproteinases 1 (TIMP-1), intercellular adhesion molecule 1 (ICAM-1), CXC motif chemokine ligand 7 (CXCL7), or CD62L cytokine were determined using the Rat Cytokine Antibody Array kit (catalog no. ARY008; R&D Systems, Minneapolis, MN, USA) following the manufacturer’s instructions. The expression levels were normalized to the value for the control.
QUANTITATIVE REAL-TIME PCR (QRT-PCR):
Total RNA was obtained from frozen rat liver using Trizol reagent (Life Technologies, Carlsbad, CA, USA), the residual genomic DNA (gDNA) was removed by incubation with RNase-free DNase, and RNA integrity was confirmed by formaldehyde gel electrophoresis. Quantification was performed using a NanoDrop1000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). Reverse-transcription of total RNA (1 µg) into cDNA was performed using oligo-dT and dNTP, and then the mRNA level was determined by RT- PCR using SYBER Green I Master (Roche Diagnostics GmbH, Mannheim, Germany). Relative changes in mRNA expression levels were determined using qRT-PCR. The cycle number at which the transcripts were detectable [Cq (Ct)] was normalized to the cycle number of GADPH mRNA detection. The primer sequences used for qRT-PCR were:
STATISTICAL ANALYSIS:
All data are expressed as mean±standard error (SE). The
Results
:
To investigate the effects of ASD on liver damage, male albino Wistar mice were exposed for 6 weeks to TAA with or without 50 mg/kg ASD. Biochemical analysis of liver enzymes showed that while TAA significantly increased production of alanine transaminase (ALT; 6.58-fold, p<0.01) and aspartate transaminase (AST; 6.05-fold, p<0.01) from baseline levels in the control group, treatment with 50 mg/kg ASD repressed the TAA-enhanced ALT or AST by 3.12-fold (p<0.01) or 2.75-fold (p<0.01), respectively (Figure 1A). Concurrently, in contrast to the unaltered body weight of rats in the control or TAA+ASD groups relative to the pre-experiment baseline median rat weights (228±3.5 g), the body weights of TAA-treated rats increased in a graduated manner during the study, attaining ~496 g by day 42 of the experiment (Figure 1B). While we observed minor significant differences in liver weight between the control rats and TAA-treated rats, the TAA+ASD-treated rats exhibited significant reduction in liver weight (1.53-fold reduction, p<0.05) compared to the TAA-treated group (Figure 1C), which is suggestive of the ameliorative effect of ASD in subjects with liver injury. Survival analysis of the rats revealed that, compared to the 100% survival rate of rats from the TAA+ASD group, survival rates on days 20 and 38 for the TAA group were reduced to 69% and 56%, respectively; while for the control group, it was 100% and 80%, respectively (Figure 1D), suggesting that ASD elicits better survival in treated rats with liver injury. These results indicate that the administration of ASD significantly attenuated extraneously-induced liver damage and facilitated survival.
ASD REVERSED TAA-INDUCED LIVER INJURY AND FIBROGENESIS IN LIVER INJURY MODEL RATS:
To assess the therapeutic effect of ASD in rats with TAA-induced liver injury, we performed extensive histological evaluation of samples derived from the model rats. H&E staining revealed that, compared to rats in the control group, liver tissues from the TAA group exhibited hepatocyte damage, with the formation and apparent expansion of fibrous septa; conversely, compared with the TAA group, the TAA+ASD rats showed significantly reduced hepatic injury and fibrosis (Figure 2A). Furthermore, using Picro Sirius red stain, we demonstrated that, compared with samples from the control or TAA+ASD group, liver samples from the TAA-treated rats showed marked increase in the population of Picro Sirius-stained collagen fibers (Figure 2B). In similar experiments, we observed significant increases in the number of α-SMA-positive liver cells around the fibrous septa, sinusoids, and portal tracts of the TAA group, compared with liver tissue from the control or TAA+ASD rats (Figure 2C). Consistent with the above, the fibrosis scores in TAA rats were 4.5-fold (p<0.01) or 3.21-fold (p<0.01) higher than in the control or TAA+ASD rats, respectively (Figure 2D), indicating that treatment with ASD significantly ameliorated TAA-induced liver fibrosis. These results were corroborated by our data showing that TAA induced a significant increase (~78-fold, p<0.001) in the collagen-positive area in the liver; conversely, this increase was suppressed by ASD treatment (~59-fold reduction, p<0.001) (Figure 2E). These data indicate that ASD treatment reversed TAA -induced liver injury and fibrogenesis.
ASD SUPPRESSED THE EXPRESSION OF α-SMA AND PRODUCTION OF OTHER FIBROGENESIS-RELATED GENES OR CYTOKINE LEVELS IN THE LIVER INJURY MODEL RATS:
Following our results indicating that ASD reversed TAA-induced liver injury, in vivo, we sought to determine the underlying mechanism. First, using Western blot analysis, we confirmed that treatment with ASD significantly suppressed TAA-enhanced expression of α-SMA protein, as demonstrated by the 2.4-fold (p<0.05) decrease in α-SMA expression in the TAA+ASD group compared with the TAA group (Figure 3A, 3B). Then, we showed that while exposure to TAA strongly concomitantly enhanced the mRNA expression of fibrosis- and ECM synthesis-related genes (α-SMA, procollagen, ICAM-1, MMP2, MMP9, and MMP13), rats in the TAA+ASD group exhibited significantly reduced ICAM-1, MMP2, MMP9, and MMP13 mRNA expression levels as determined by qRT-PCR (Figure 3C). Similarly, assessment of the effect of ASD on circulating levels of cytokine mediators of ECM formation using an ELISA-based rat cytokine array showed that treatment with ASD significantly reduced TIMP-1 (0.9-fold, p<0.05), ICAM-1 (1.75-fold, p<0.01), CXCL7 (1.74-fold, p<0.01), and CD62L (1.2-fold, p<0.01) cytokine levels in the TAA+ASD group compared with their TAA counterparts (Figure 3D). Taken together, these results indicate that ASD suppressed the production of TAA-induced fibrogenic and ECM synthesis cytokines in the serum and their expression in injured liver tissues.
ASD AMELIORATED OXIDATIVE STRESS VIA MODULATION OF NRF2/JUND/NF-κB P65 SIGNALING IN RATS WITH TAA-INDUCED LIVER INJURY:
We sought to further define the mechanism underlying the pharmacologic activity of ASD. Because of the critical role of Nrf-2 in the induction of stress-inducible genes, involvement of Nrf2/NF-κB p65 signaling in the induction and severity of inflammatory processes [12], as well as the implication of JunD in inflammation and fibrogenesis [13,14], we evaluated the probable effect of ASD on Nrf2, JunD, and NF-κB p65 signaling in the TAA-induced liver injury model rats using Western blot and biochemical analyses. Our Western blot analysis data showed that, compared to the TAA group, there was significantly suppressed protein expression of nuclear Nrf2 and JunD in the TAA+ASD liver cells, similar to baseline levels in the control group; similarly, but to a lesser extent, expression of NF-κB p65 protein was only moderately downregulated (Figure 4A). Quantitatively, compared to the TAA group, we demonstrated a 1.5-fold (p<0.01), 1.2-fold (p<0.01), and 1.61-fold (p<0.01) decrease in Nrf-2, NF-κB p65, and JunD proteins expression, respectively, in the livers of ASD-treated rats (Figure 4B). These data show the efficacy of ASD in amelioration of hepatic oxidative stress. In addition, in validation tests based on quantification of total glutathione (GSH) and its oxidized variant glutathione disulfide (GSSG), we demonstrated that ASD effectively reversed TAA-induced increase in the GSH/GSSG ratio (0.4-fold difference, p<0.05), almost back to baseline level seen in the control group (Figure 4C),suggesting a role for ASD in the restoration of the reduced GSH levels in the rat livers. This trend was replicated with markers of lipid peroxidation 4-Hydroxynonenal (4-HNE) and malondialdehyde (MDA), which are indicators of oxidative stress. Our results showed that, similar to the control group, but in contrast to the TAA group, production of 4-HNE and MDA in the TAA+ASD group was suppressed by 1.5-fold (p<0.01) and 1.1-fold (p<0.05), respectively (Figure 4D, 4E).
Discussion
The present study reports for the first time, to the best of our knowledge, the therapeutic effects of ASD on TAA-induced liver fibrosis and the probable underlying molecular mechanism in a rat model. Using TAA-induced rat liver fibrosis models, we demonstrated significantly enhanced production of liver function biomarkers, ALT, and AST in the TAA group compared to the control or ASD-treated group, and most rats in the TAA group gained weight.
Rats in the TAA+ASD group, after treatment with the ASD, exhibited improved liver function enzymes production compared to rats in the TAA group (
The present study also provided insight into the probable mechanisms underlying the therapeutic activity of ASD in subjects with inflammation-associated liver fibrosis and/or impaired liver function. The inflammatory cascade is broadly implicated in the induction and/or augmentation of stress-related organ injury [17–19]. This is consistent with our findings of concomitantly enhanced production of the pro-inflammatory NF-κB p65 and oxidative stress markers Nrf-2 and JunD in the rat model of TAA-induced liver injury, which was effectively inhibited by treatment with ASD (Figure 4), indicating that the ameliorative effect of ASD in the pre-clinical models of TAA-induced liver injury is probably mediated by the suppression of inflammation-related oxidative stress. This would particularly be so because JunD rescues increased oxidative stress and expression of Nrf1 and Nrf2 in Jun(d/d) fetal livers [20], and because TAA-induced toxicity and liver injury are associated with enhanced release of free radicals and oxidative stress [21]. Furthermore, the ubiquitous tripeptide thiol GSH has been shown to play a critical role in the detoxification of xenobiotics and heavy metals, and is an essential intra- and extracellular antioxidant, and the depletion of the antioxidant GSH is one of the indices of oxidative stress [22]. It is thus interesting that ASD significantly reversed the TAA-induced reduction of the GSH/GSSG ratio, restoring it to near baseline level (Figure 4), especially as highly reduced GSH levels have been linked with enhanced oxidative damage by free radicals in liver injury models. Consistently, the TAA-induced enhanced production of lipid peroxidation proteins 4-HNE and MDA was markedly decreased in the ASD-treated rats. These results indicate that ASD ameliorates TAA-induced injury by reducing cellular oxidation.
Conclusions
Our findings indicate that ASD exhibits strong hepato-protective and reparative effects, as it alleviates TAA-induced hepatic oxidative stress and deactivates the Nrf-2/JunD/NF-κB signaling axis (Figure 5). The present study provides new insights into the hepato-protective effects of ASD.
Figures
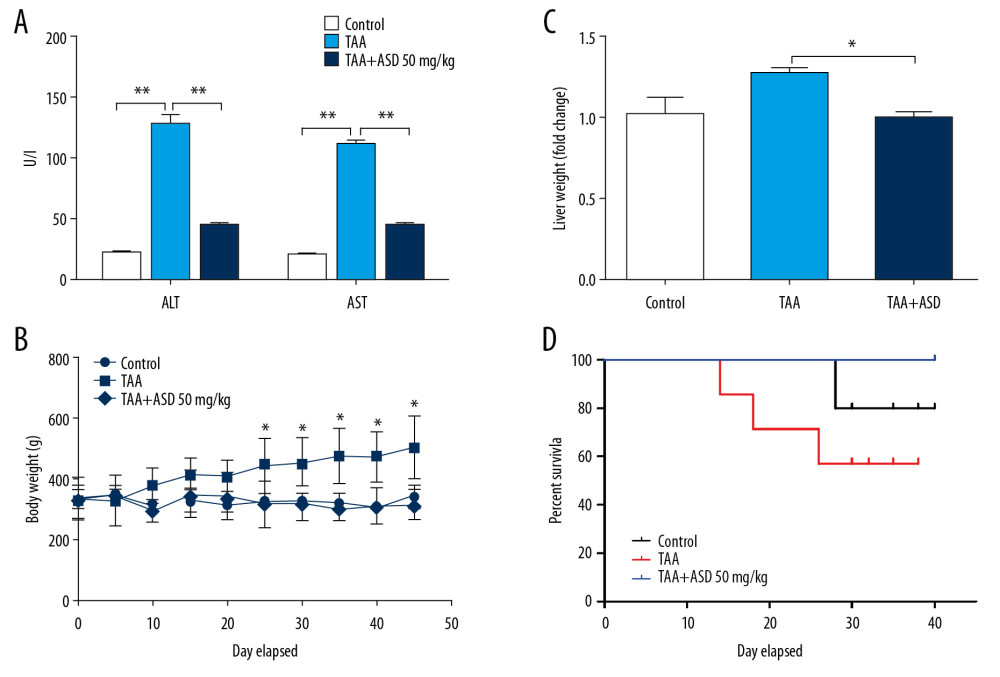 Figure 1. ASD ameliorated thioacetamide-induced hepatotoxicity and associated hepatic dysfunction in vivo. Graphical representations of the effect of 50 mg/kg ASD on (A) TAA-induced ALT or AST elevation, (B) rat liver weight, and (C) rat body weight. (D) Kaplan-Meier plot of the effect of 50 mg/kg ASD on the rat survival rate. TAA – thioacetamide; ASD – Alisma Shugan Decoction; ALT – alanine transaminase; AST – aspartate transaminase. * p<0.05; ** p<0.01; *** p<0.001.
Figure 1. ASD ameliorated thioacetamide-induced hepatotoxicity and associated hepatic dysfunction in vivo. Graphical representations of the effect of 50 mg/kg ASD on (A) TAA-induced ALT or AST elevation, (B) rat liver weight, and (C) rat body weight. (D) Kaplan-Meier plot of the effect of 50 mg/kg ASD on the rat survival rate. TAA – thioacetamide; ASD – Alisma Shugan Decoction; ALT – alanine transaminase; AST – aspartate transaminase. * p<0.05; ** p<0.01; *** p<0.001. 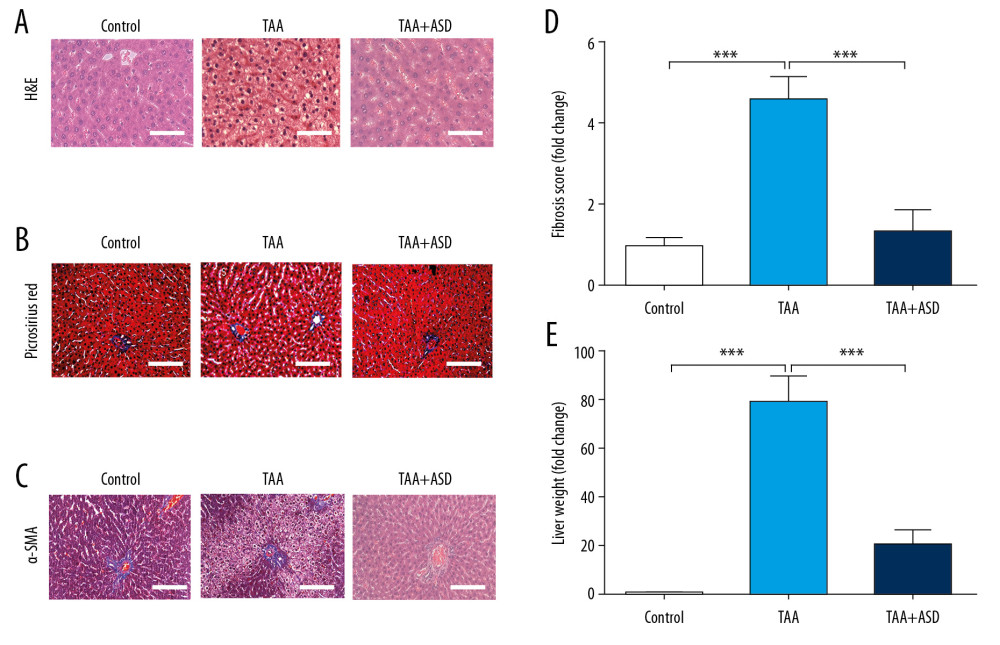 Figure 2. ASD reversed TAA-induced liver injury and fibrogenesis in liver injury model rats. (A) Images showing the effect of TAA alone or with ASD on the liver morphohistology in the liver injury model rats. Images of the effect of TAA alone or with ASD on the (B) picrosirius red-stained collagen fibers, and (C) α-SMA expression. Graphs showing how TAA alone or with ASD affected the (D) fibrosis score and (E) collagen area in the liver injury model rats. *** p<0.001.
Figure 2. ASD reversed TAA-induced liver injury and fibrogenesis in liver injury model rats. (A) Images showing the effect of TAA alone or with ASD on the liver morphohistology in the liver injury model rats. Images of the effect of TAA alone or with ASD on the (B) picrosirius red-stained collagen fibers, and (C) α-SMA expression. Graphs showing how TAA alone or with ASD affected the (D) fibrosis score and (E) collagen area in the liver injury model rats. *** p<0.001. 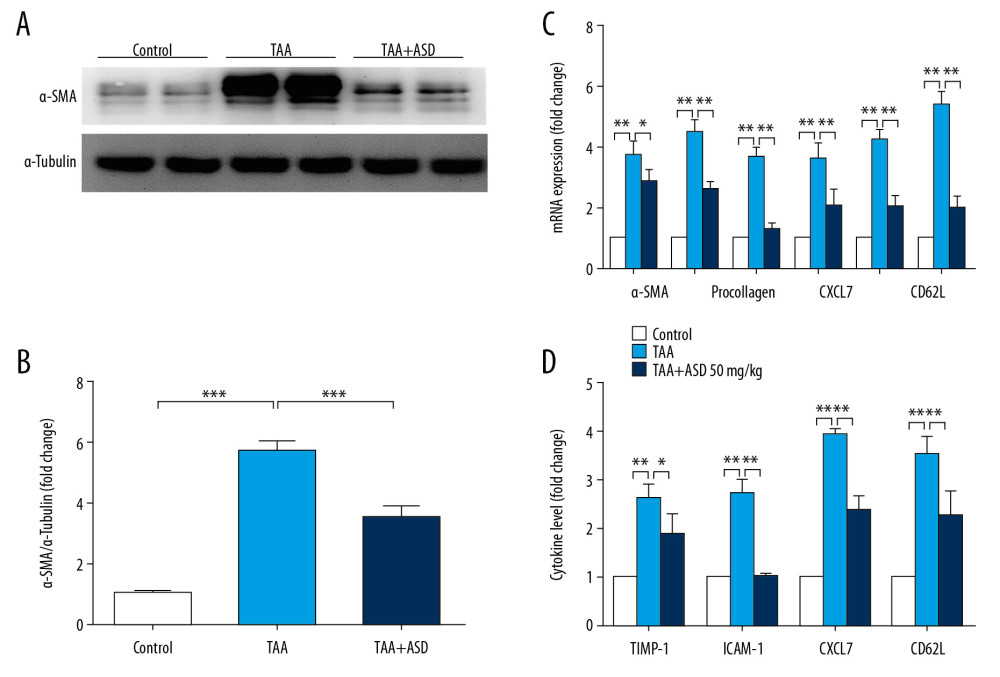 Figure 3. ASD suppressed the expression of α-SMA and production of other fibrogenesis-related genes or cytokine levels in the liver injury model rats. (A) Western blot image and (B) graph showing the effect of ASD on the expression of α-SMA protein in cells from rats with TAA-induced liver injury. Histograms of the effect of ASD on the (C) expression levels of α-SMA, procollagen, ICAM-1, MMP2, MMP9, and MMP13 mRNAs, or (D) TIMP-1, ICAM-1, CXCL7, and CD62L cytokine levels in cells from rats with TAA-induced liver injury. α-tubulin was used as loading and internal control. * p<0.05; ** p<0.01; *** p<0.001.
Figure 3. ASD suppressed the expression of α-SMA and production of other fibrogenesis-related genes or cytokine levels in the liver injury model rats. (A) Western blot image and (B) graph showing the effect of ASD on the expression of α-SMA protein in cells from rats with TAA-induced liver injury. Histograms of the effect of ASD on the (C) expression levels of α-SMA, procollagen, ICAM-1, MMP2, MMP9, and MMP13 mRNAs, or (D) TIMP-1, ICAM-1, CXCL7, and CD62L cytokine levels in cells from rats with TAA-induced liver injury. α-tubulin was used as loading and internal control. * p<0.05; ** p<0.01; *** p<0.001. 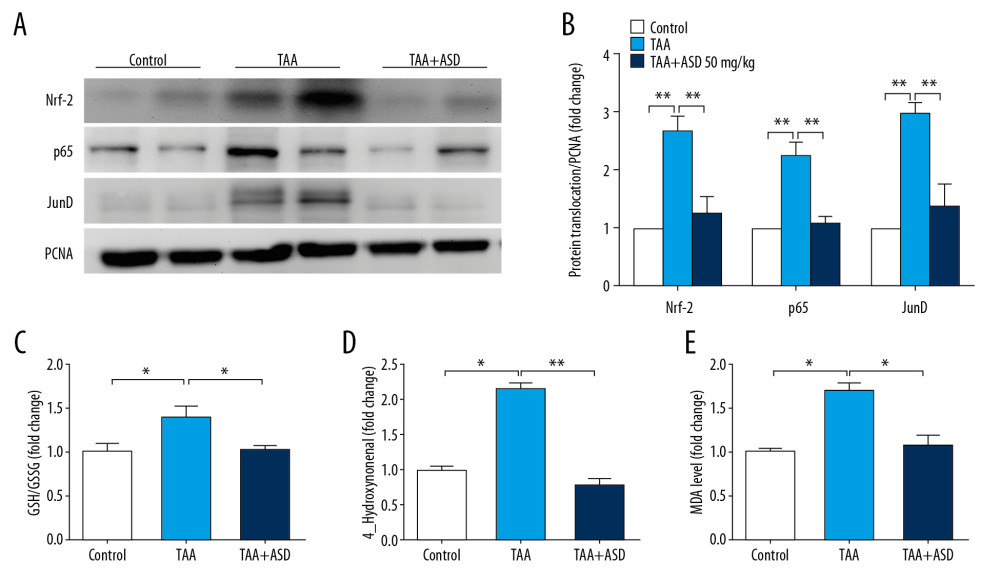 Figure 4. ASD ameliorated oxidative stress via modulation of Nrf2/JunD/p65 signaling in rats with TAA-induced liver injury. (A) Representative Western blot images and (B) histograms showing the effect of TAA or TAA+ASD on the expression of nuclear-translocated Nrf2, p65, and JunD in rats with TAA-induced liver injury. Histograms of the effect of TAA or TAA+ASD on the (C) GSH/GSSG ratio, (D) 4-hydroxynonenal, or (E) MDA levels in the liver injury model rats. PCNA was used as nuclear marker and as loading control. PCNA – proliferating cell nuclear antigen; GSH – glutathione; GSSG – glutathione disulfide; MDA – malondialdehyde. * p<0.05; ** p<0.01; *** p<0.001.
Figure 4. ASD ameliorated oxidative stress via modulation of Nrf2/JunD/p65 signaling in rats with TAA-induced liver injury. (A) Representative Western blot images and (B) histograms showing the effect of TAA or TAA+ASD on the expression of nuclear-translocated Nrf2, p65, and JunD in rats with TAA-induced liver injury. Histograms of the effect of TAA or TAA+ASD on the (C) GSH/GSSG ratio, (D) 4-hydroxynonenal, or (E) MDA levels in the liver injury model rats. PCNA was used as nuclear marker and as loading control. PCNA – proliferating cell nuclear antigen; GSH – glutathione; GSSG – glutathione disulfide; MDA – malondialdehyde. * p<0.05; ** p<0.01; *** p<0.001. 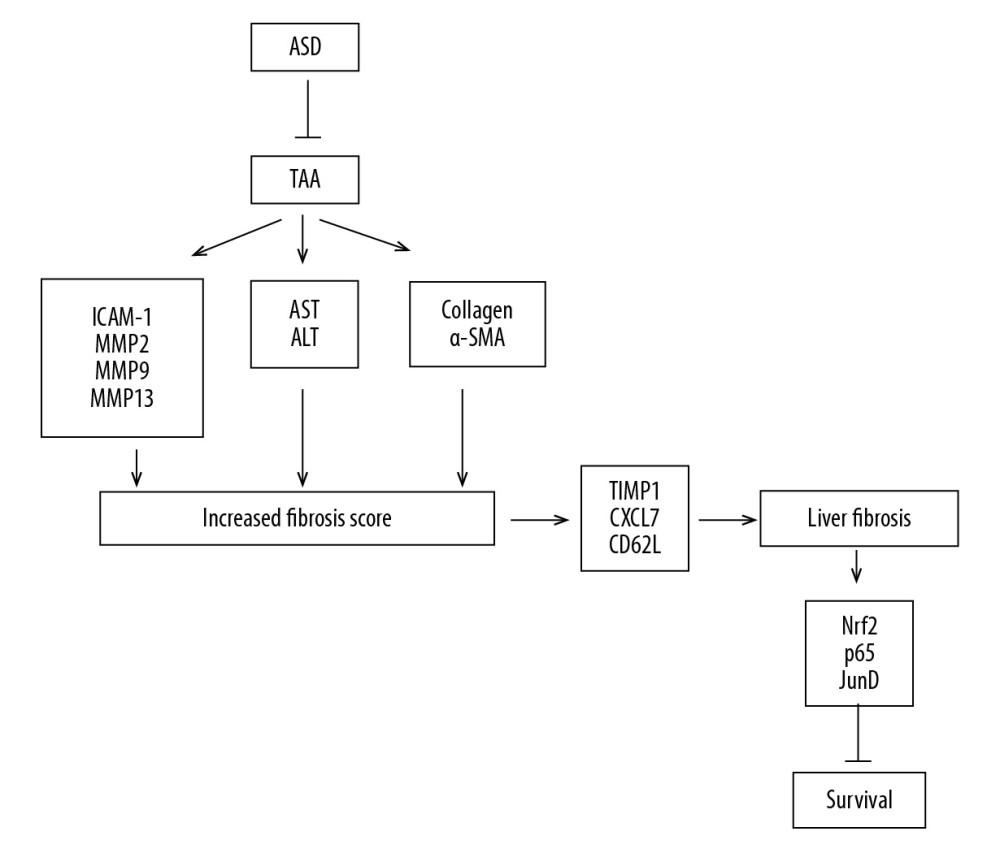 Figure 5. Pictorial abstract showing how Alisma Shugan Decoction (ASD) ameliorated hepatotoxicity and associated liver dysfunction. The present study provides new insights into the hepato-protective effects of ASD.
Figure 5. Pictorial abstract showing how Alisma Shugan Decoction (ASD) ameliorated hepatotoxicity and associated liver dysfunction. The present study provides new insights into the hepato-protective effects of ASD. 
References
1. Bataller R, Brenner DA, Liver fibrosis: J Clin Invest, 2005; 115(2); 209-18 Erratum in: J Clin Invest, 2005; 115(4): 1100
2. Elpek GO, Cellular and molecular mechanisms in the pathogenesis of liver fibrosis: An update: World J Gastroenterol, 2014; 20(23); 7260-76
3. Seki E, Brenner DA, Recent advancement of molecular mechanisms of liver fibrosis: Hepatobiliary Pancreat Sci, 2015; 22(7); 512-18
4. Mallat A, Lotersztajn S, Cellular mechanisms of tissue fibrosis. 5. Novel insights into liver fibrosis: Am J Physiol Cell Physiol, 2013; 305(8); C789-99
5. Kisseleva T, Brenner DA, Anti-fibrogenic strategies and the regression of fibrosis: Best Pract Res Clin Gastroenterol, 2011; 25(2); 305-17
6. Koyama Y, Brenner DA, New therapies for hepatic fibrosis: Clin Res Hepatol Gastroenterol, 2015; 39(Suppl 1); S75-79
7. Pinzani M, Pathophysiology of liver fibrosis: Dig Dis, 2015; 33(4); 492-97
8. Sánchez-Valle V, Chávez-Tapia NC, Uribe M, Méndez-Sánchez N, Role of oxidative stress and molecular changes in liver fibrosis: A review: Curr Med Chem, 2012; 19(28); 4850-60
9. Luangmonkong T, Suriguga S, Mutsaers HAM, Targeting oxidative stress for the treatment of liver fibrosis: Rev Physiol Biochem Pharmacol, 2018; 175; 71-102
10. Zhao CQ, Zhou Y, Ping J, Xu LM, Traditional Chinese medicine for treatment of liver diseases: Progress, challenges and opportunities: J Integr Med, 2014; 12; 401-8
11. Ishak K, Baptista A, Bianchi L, Histological grading and staging of chronic hepatitis: J Hepatol, 1995; 22(6); 696-99
12. Ahmed SM, Luo L, Namani A, Nrf2 signaling pathway: Pivotal roles in inflammation: Biochim Biophys Acta Mol Basis Dis, 2017; 1863(2); 585-97
13. Carr TM, Wheaton JD, Houtz GM, Ciofani M, JunB promotes Th17 cell identity and restrains alternative CD4+ T-cell programs during inflammation: Nat Commun, 2017; 8(1); 301
14. Smart DE, Green K, Oakley F, JunD is a profibrogenic transcription factor regulated by Jun N-terminal kinase-independent phosphorylation: Hepatology, 2006; 44(6); 1432-40
15. Kisseleva T, The origin of fibrogenic myofibroblasts in fibrotic liver: Hepatology, 2017; 65(3); 1039-43
16. Iredale JP, Thompson A, Henderson NC, Extracellular matrix degradation in liver fibrosis: Biochemistry and regulation: Biochim Biophys Acta Mol Basis Dis, 2013; 1832(7); 876-83
17. Liu YZ, Wang YX, Jiang CL, Inflammation: The common pathway of stress-related diseases: Front Hum Neurosci, 2017; 11; 316
18. Chen L, Deng H, Cui H, Inflammatory responses and inflammation-associated diseases in organs: Oncotarget, 2017; 9(6); 7204-18
19. Polimeni L, Del Ben M, Baratta F, Oxidative stress: New insights on the association of non-alcoholic fatty liver disease and atherosclerosis: World J Hepatol, 2015; 7(10); 1325-36
20. Meixner A, Karreth F, Kenner L, Jun and JunD-dependent functions in cell proliferation and stress response: Cell Death Differ, 2010; 17(9); 1409-19
21. Oliver JR, Jiang S, Cherian MG, Augmented hepatic injury followed by impaired regeneration in metallothionein-I/II knockout mice after treatment with thioacetamide: Toxicol Appl Pharmacol, 2006; 210(3); 190-99
22. Zitka O, Skalickova S, Gumulec J, Redox status expressed as GSH: GSSG ratio as a marker for oxidative stress in paediatric tumour patients: Oncol Lett, 2012; 4(6); 1247-53
Figures
Figure 1. ASD ameliorated thioacetamide-induced hepatotoxicity and associated hepatic dysfunction in vivo. Graphical representations of the effect of 50 mg/kg ASD on (A) TAA-induced ALT or AST elevation, (B) rat liver weight, and (C) rat body weight. (D) Kaplan-Meier plot of the effect of 50 mg/kg ASD on the rat survival rate. TAA – thioacetamide; ASD – Alisma Shugan Decoction; ALT – alanine transaminase; AST – aspartate transaminase. * p<0.05; ** p<0.01; *** p<0.001.Figure 2. ASD reversed TAA-induced liver injury and fibrogenesis in liver injury model rats. (A) Images showing the effect of TAA alone or with ASD on the liver morphohistology in the liver injury model rats. Images of the effect of TAA alone or with ASD on the (B) picrosirius red-stained collagen fibers, and (C) α-SMA expression. Graphs showing how TAA alone or with ASD affected the (D) fibrosis score and (E) collagen area in the liver injury model rats. *** p<0.001.Figure 3. ASD suppressed the expression of α-SMA and production of other fibrogenesis-related genes or cytokine levels in the liver injury model rats. (A) Western blot image and (B) graph showing the effect of ASD on the expression of α-SMA protein in cells from rats with TAA-induced liver injury. Histograms of the effect of ASD on the (C) expression levels of α-SMA, procollagen, ICAM-1, MMP2, MMP9, and MMP13 mRNAs, or (D) TIMP-1, ICAM-1, CXCL7, and CD62L cytokine levels in cells from rats with TAA-induced liver injury. α-tubulin was used as loading and internal control. * p<0.05; ** p<0.01; *** p<0.001.Figure 4. ASD ameliorated oxidative stress via modulation of Nrf2/JunD/p65 signaling in rats with TAA-induced liver injury. (A) Representative Western blot images and (B) histograms showing the effect of TAA or TAA+ASD on the expression of nuclear-translocated Nrf2, p65, and JunD in rats with TAA-induced liver injury. Histograms of the effect of TAA or TAA+ASD on the (C) GSH/GSSG ratio, (D) 4-hydroxynonenal, or (E) MDA levels in the liver injury model rats. PCNA was used as nuclear marker and as loading control. PCNA – proliferating cell nuclear antigen; GSH – glutathione; GSSG – glutathione disulfide; MDA – malondialdehyde. * p<0.05; ** p<0.01; *** p<0.001.Figure 5. Pictorial abstract showing how Alisma Shugan Decoction (ASD) ameliorated hepatotoxicity and associated liver dysfunction. The present study provides new insights into the hepato-protective effects of ASD. In Press
Clinical Research
Effects of Single-Bout Endurance Exercise Intensity on Peripheral Neurotrophic Factors in Patients With Isc...Med Sci Monit In Press; DOI: 10.12659/MSM.952089
Review article
Anisodus tanguticus in Cancer Research: A Review of Traditional Use, Phytochemistry, Extraction Methods, an...Med Sci Monit In Press; DOI: 10.12659/MSM.952999
Clinical Research
Nasal Mucociliary Clearance and Its Relationship With Disease Severity in Patients With Multiple SclerosisMed Sci Monit In Press; DOI: 10.12659/MSM.952850
Clinical Research
Modified Thoracoabdominal Nerves Block Through the Perichondrial Approach vs Subcostal Transversus Abdomini...Med Sci Monit In Press; DOI: 10.12659/MSM.953976
Most Viewed Current Articles
17 Jan 2024 : Review article 14,176,570
Vaccination Guidelines for Pregnant Women: Addressing COVID-19 and the Omicron VariantDOI :10.12659/MSM.942799
Med Sci Monit 2024; 30:e942799
13 Nov 2021 : Clinical Research 3,762,188
Acceptance of COVID-19 Vaccination and Its Associated Factors Among Cancer Patients Attending the Oncology ...DOI :10.12659/MSM.932788
Med Sci Monit 2021; 27:e932788
14 Dec 2022 : Clinical Research 2,466,310
Prevalence and Variability of Allergen-Specific Immunoglobulin E in Patients with Elevated Tryptase LevelsDOI :10.12659/MSM.937990
Med Sci Monit 2022; 28:e937990
16 May 2023 : Clinical Research 708,927
Electrophysiological Testing for an Auditory Processing Disorder and Reading Performance in 54 School Stude...DOI :10.12659/MSM.940387
Med Sci Monit 2023; 29:e940387