05 September 2020: Database Analysis
Identification of Hub Genes Associated with Hypertension and Their Interaction with miRNA Based on Weighted Gene Coexpression Network Analysis (WGCNA) Analysis
Zongjin Li1ABCDEF, Jacqueline Chyr2EF, Zeyu Jia3BF, Lina Wang4EF, Xi Hu4DF, Xiaoming Wu4AC, Changxin Song5DG*DOI: 10.12659/MSM.923514
Med Sci Monit 2020; 26:e923514






Abstract
BACKGROUND: Hypertension is one of the most widespread health conditions in the world, and the molecular mechanism of it is still unclear. In this study, we identified the hub genes (hub miRNA genes) associated with hypertension and explored the relationship between hypertension miRNA-gene by constructing a mRNA co-expression network and a miRNA co-expression network, which can help to reveal the mechanism and predict the prognosis of hypertension progression.
MATERIAL AND METHODS: Based on gene expression profile data of hypertensive samples from the Gene Expression Omnibus database, WGCNA was used to detect hypertension-related biomarkers and key mRNA and miRNA modules. Then, DAVID was used to perform gene-annotation enrichment analysis and Kyoto Encyclopedia of Genes and Genomes (KEGG) and miRPath were used for pathway analysis of mRNA and miRNAs genes.
RESULTS: We identified 3 key modules relating to hypertension, 2 mRNA modules named Msaddlebrown and Mgreenyellow and 1 miRNA module named Msalmon. In addition, 12 hub genes (RPL21, RPS28, LOC442727/PTGAP10, LOC100129599/RPS29P14, TBXAS1, FCER1G, CFP, FURIN, PECAM1, IGSF6, NCF1C, and LOC285296/UNC93B3) and 7 hub miRNAs (hsa-miR-1268a/b, hsa-miR-513c-3p, hsa-miR-4799-5p, hsa-miR-296-3p, hsa-miR-5195-5p, hsa-miR-219-2-3p, and hsa-miR-548d-5p) relating to hypertension were identified. HIF-1 signaling pathway and insulin signaling pathway were closely related to the 3 key modules. We also discovered 4 miRNAs (hsa-miR-548am-3p, hsa-miR-513c-3p, hsa-miR-182-5p, and hsa-miR-548d-5p) and 6 genes (IGF1R, GSK3B, FOXO1, PRKAR2B, HIF1A, and PIK3R1) were the core nodes in the hypertension-related miRNA-gene network, and hsa-miR-548am-3p was at the center of the network.
CONCLUSIONS: These findings will help improve the understanding of the pathogenesis of hypertension, and the discovered genes can serve as signatures for early diagnosis of hypertension.
Keywords: Biological Markers, Hypertension, Computational Biology, Gene Regulatory Networks
Background
Hypertension, also known as high blood pressure, is one of the most widespread health conditions in the world. It is one of the most dangerous factors affecting cardiovascular death. Every year, more than 17 million people die from cardiovascular disease. Hypertension is responsible for more than 45% of deaths due to heart disease, and 51% of deaths due to stroke [1]. The risk factors for hypertension include age, ethnicity, weight, diet, alcohol and tobacco use, gender, as well as existing health condition such as diabetes, high cholesterol levels, and chronic kidney disease. The pathogenic mechanism is even more complex, involving a variety of molecules and pathways [2].
Currently, hypertensive patients are treated with drugs such as diuretics, vasodilators, rapid-acting intravenous antihypertensive agents, and other drugs. Unfortunately, hypertension medications have many side effects such as cough, diarrhea, dizziness, drowsiness, depression, ulcers, and more. Since hypertension is a multiple-factor disease, further research is needed to reveal the molecular mechanism of hypertension and new biomarkers need to be discovered. The increased availability of high-throughput sequencing and microarrays along with the development of bioinformatics tools and algorithms has allowed for the discovery of several biomarkers that regulate blood pressure. For example, the anti-inflammatory cytokine interleukin-10 (IL-10) has a strong antihypertensive effect [3];
The network of interactions between biomolecules provides an important basis for systematic research on disease. WGCNA is an R package, which is based on the similarity between genes to construct a weighted correlation network [11]. It has unique advantages in handling complex data with multiple samples, which is a powerful method to uncover basic mechanism of gene–disease relationships [12]. Using WGCNA, Zhang et al. discovered ten hub genes that could be used as biomarkers for oral squamous cell carcinoma tumors [13]. In another study, six hub genes were found to regulate the signaling pathway of clear cell renal cell carcinoma (ccRCC) [14]. Wu et al. applied WGCNA to the identification of potential therapeutic targets for angiotensin II (Ang II) induced hypertension [7]. Here, we utilized the WGCNA method to construct gene and miRNA modules, identified new biomarkers related to hypertension, and explored the relationship between hypertension and marker genes.
Material and Methods
DATA SOURCES AND PREPROCESSING:
GSE75360, GSE75670, and GSE117261 datasets were downloaded from Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo/). GSE75360 contained 10 hypertensive and 11 normal human gene expression data, (Illumina HumanHT-12 v.4.0 Expression BeadChip). GSE75670 contained 6 hypertensive and 6 normal human miRNA expression data (Exiqon mercury™ LNA™ microRNA array, 7th generation [miRbase v18]). GSE117261 contained 58 pulmonary arterial hypertension and 25 normal human gene expression data ([HuGene-1_0-st] Affymetrix Human Gene 1.0 ST Array [transcript (gene) version]). Data preprocessing procedures such as correction of expression matrix, quality evaluation of expression data, and sample clustering were conducted in R version 3.5.2. Network analysis with WGCNA software package was also conducted in R version 3.5.2. The overall analysis workflow is showed in Figure 1.
MODULE CONSTRUCTION BASE ON WGCNA ALGORITHM:
WGCNA explores the complex relationships between genes and phenotypes by constructing scale-free co-expression networks. It transforms gene expression data into co-expression modules and then identifies hub genes in the modules. In this research, we constructed 2 weighted co-expression networks based on mRNA and miRNA expression data separately. The construction processes were the same except that some parameter values were different, so we only introduced the construction process of gene co-expression network. First, the correlation of all gene pairs was calculated to construct a similarity matrix. Second, the soft threshold β was a weighted parameter of the adjacent function, which the optimal value was obtained by the pickSoftThreshold function in the R package WGCNA [15]. Third, the TOM similarity function was used to convert the adjacency value into a TOM matrix. Then, using dissimilarity matrix dissTOM=1-TOM, we clustered the genes into the hierarchy to get the system clustering tree [13]. Fourth, mRNAs with similar expression profile were divided to the same module. According to the number of the genes and miRNAs, the minModuleSize of the mRNA was set to 50 and the minModuleSize of the miRNA was set to 30 [16]. Finally, we calculated the differences of the modules eigengenes, and set an appropriate cutline for the modules dendrogram to merge highly similar modules.
IDENTIFICATION OF KEY MODULES IN CO-EXPRESSED NETWORKS:
Two methods were used to identify key modules related to hypertension. The first method was to calculate the Pearson correlation coefficient and significance P-value of module eigengenes (MEs) and hypertension trait. Here, MEs represents the overall expression level of the gene module [17]. The second method calculates the gene significance (GS) and the module significance (MS). Here, GS is the correlation between a gene and the clinical features. MS is the average GS of all genes in a module [13]. Generally, the higher the absolute value of MS and GS, the more relevant the gene module is to hypertension.
ENRICHMENT ANALYSIS OF KEY MODULES:
To further understand the function of a key module and its biological significance, we used the online functional annotation database DAVID (https://david.ncifcrf.gov/). For the mRNA modules, we used Gene Ontology (GO) terms and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis to identify significantly enriched pathways (P-value <0.05). For miRNA modules, the online database miRPath V.3 (http://snf-515788.vm.okeanos.grnet.gr/) was used to predict miRNAs target genes for KEGG pathway enrichment [18].
IDENTIFY HUB GENES AND HUB MIRNAS:
This study used 2 methods to identify hub genes (miRNA) from key modules: 1) Importance threshold, and 2) MCC (maximal clique centrality function) algorithm which used the cytoHubba plugin. In the first method, hub genes are defined as genes with high GS value, high modular membership (MM) value, and low weighted P-value associated with genes and hypertension (P. weighted). MM was used to measure the importance of genes in modules [19]. The P-weighted value was calculated by the networkScreening function in WGCNA. A P. weighted value less than 0.05 was considered biologically significant [20]. In the second method, we used the MCC algorithm in cytoHubba plug-in from the Cytoscape software to identify hub genes [21].
GENE SELECTION AND HUB GENE VERIFICATION:
We used limma R package to analyze differently expressed genes (DEGs) between normal samples and hypertension samples in the dataset GSE75360 and set the cutoff value to log2FC< |0.182| and P-value <0.05. The volcanic map and hierarchical clustering analysis were performed by “ggplot2” and “pheatmap” package of R, respectively. Then, we used the jvenn (http://jvenn.toulouse.inra.fr) to draw Venn diagrams to overlap the genes in DEGs and hub genes [22]. We verified our hub genes using a different DEG dataset (GSE117261). We queried the role of hub genes through The Human Protein Atlas database (https://www.Proteinatlas.org/) and used NCBI (https://www.ncbi.nlm.nih.gov/gene/) to verify whether these hub genes could be used as biomarkers of hypertension.
CONSTRUCTION OF MIRNA-GENE INTERACTIVE NETWORK:
DAVID and miRPath were used to identify enriched mRNA and miRNA pathways, respectively. Then, the overlapping pathways were chosen for network construction. Cytoscape was used to construct miRNA-gene interaction networks.
Results
DATA PREPROCESSING:
After preprocessing, an expression matrix containing 29 595 genes was obtained. We calculated standard deviation (SD) of all genes, then, ranked the SD from large to small, and selected the top 6000 genes as the input data for the construction of the gene expression network. We performed the same preprocessing procedures on the miRNA expression matrix. A total of 1916 miRNAs were selected for co-expression network construction of miRNAs.
CONSTRUCTION OF WEIGHTED CO-EXPRESSION NETWORK:
To obtain a network that meets the scale-free topology criterion, we calculated network structures by using different soft-thresholding power range from 1 to 20. The scale-free topological fitting index of mRNA co-expression network reaches 0.8, when the soft-thresholding power was 14 (Supplementary Figure 1A, 1B), which met the scale-free network criterion. Then, the genes were clustered into modules by hierarchical clustering according to expression value, and the most similar modules were merged by setting the MEDissThres cutting line to 0.2 (Supplementary Figure 1C). Finally, 19 mRNA gene modules were identified (Figure 2). For miRNA co-expression networks, the soft-thresholding power was set at 9 and the scale-free topological fitting index at 0.88 (Supplementary Figure 2A, 2B). The MEDissThres cutting line was set to 0.1 (Supplementary Figure 2C), and 14 miRNA gene modules were identified (Figure 3). The relationship for each mRNA module was analyzed by drawing a network heat map (Supplementary Figure 3). The network heat map was produced with the modules in the miRNA co-expression network (Figure 4). The different colors in the vertical and horizontal axes stand for different miRNA modules. The yellow color in the middle area indicates a degree of connection for each miRNA module. The figure shows no significantly differences in the interaction between the modules, indicating that the miRNA modules had relatively high independence.
IDENTIFYING OF KEY MODULES:
We used 2 methods to identify key modules. The first method calculate the Pearson correlation and significance P-value between MEs and hypertension, and the other method calculates GS. We found 2 mRNA modules with significant correlations to hypertension, named Msaddlebrown and Mgreenyellow. Compared to other mRNA modules, the correlation coefficient of theese two modules were the largest (Figure 5A). In the miRNA co-expression network, the Msalmon module was highly related to hypertension (Figure 5B). In order to ensure the identified modules were significantly associated with hypertension, we calculated the GS in the modules and verify the key modules again (Figure 6A, 6B). For the mRNA co-expression network, the Msaddlebrown and Mgreenyellow modules had the largest GS scores at 0.451 and 0.410, respectively. For miRNA co-expression networks, the absolute GS value of Msalmon was 0.398. These GS values all indicate that these modules were significantly associated with hypertension.
ENRICHMENT ANALYSIS OF KEY MODULES:
We performed GO analysis and KEGG analysis on the genes in the Msaddlebrown and Mgreenyellow modules to explore their biological significance (Supplementary Tables 1, 2). The Msaddlebrown module was related to RNA transcriptional translation, such as SRP-dependent co-translational protein targeting to membrane, translational initiation, and RNA binding. The Mgreenyellow module was involved in a variety of immune and metabolic processes, such as positive regulation of IL-6 production, positive regulation of transcription from RNA polymerase II promoter, glycoside catabolic process, and so on. Furthermore, KEGG pathway analysis showed enrichment in ribosome, HIF-1 signaling pathway, and osteoclast differentiation pathways.
The significant pathways in Msalmon module were Hippo signaling pathway, adherens junction, and proteoglycans in cancer (Supplementary Table 3). Among them, the HIF-1 signaling pathway and the insulin signaling pathway were shared by both mRNA and miRNA modules.
IDENTIFICATION OF HUB GENES AND MIRNAS:
According to the definition of module connectivity, we calculated the MM and GS of the genes (miRNAs) in each of the key modules to select the hub genes (miRNAs). Then, we used the networkScreening function to obtain the P. weighted of each gene (miRNA). |MM| >0.8, |GS| >0.2 and P. weighted <0.05 were used as the identification criteria. Finally, 26 genes were obtained in the Msaddlebrown module (Supplementary Table 4), 53 genes were obtained in the Mgreenyellow module (Supplementary Table 5), and 22 miRNAs were obtained in the Msalmon module (Supplementary Table 6). We also import the files of these 3 key modules into Cytoscape software and used the plugin cytoHubba to identify and visualize pivotal genes and miRNAs (Supplementary Figure 4A–4C). We finally determine that there were 4 hub genes in Msaddlebrown module, 8 hub genes in Mgreenyellow module, and 7 hub miRNAs in Msalmon module (Table 1 and Supplementary Figure 5A–5C).
THE VERIFICATION AND FUNCTIONAL ANALYSIS OF HUB GENES:
Since Msaddlebrown and Mgreenyellow modules were negatively correlated with hypertension status, we wondered if the hub genes in these 2 modules were also negatively correlated with hypertension. We verified this by correlating hub genes expression values with hypertension status. There was a significant difference in hub genes (miRNAs) expression levels between normal and hypertensive group Supplementary Figure 6A–6C. We detected the expression of 12 hub genes by DEGs on original dataset GSE75360. Figure 7 shows the DEGs in a volcano map. The hierarchical clustering heat map of DEGs is shown in Supplementary Figure 7. We used the jvenn tool to overlap the hub genes in the Msaddlebrown and Mgreenyellow module and DEGs, respectively, and found 11 hub genes were both in the DEGs gene list and the hub genes lists (Supplementary Figure 8A, 8B). We used the same method to verify the hub gene in another dataset GSE117261 from the GEO database, and found TBXAS1, FCER1G, and IGSF6 were in the DEGs gene list and hub genes lists (Supplementary Figure 9). We compared the expression status of these three hub genes in normal and hypertensive patients from the GSE117261 dataset, and the results were consistent with those from the GSE75360 dataset (Supplementary Figure 10).
ANALYSIS OF MIRNA-GENE INTERACTION NETWORKS:
We discovered that the HIF-1 signaling pathway and the insulin signaling pathway were also found in the miRNA networks. In order to better understand the regulatory relationship between genes and miRNAs, the miRNA-gene interaction network was constructed, which was based on genes and miRNAs involving the same pathway. There was a total of 46 nodes (21 genes and 25 miRNAs) and 112 pairs of interactions in the miRNA-gene interaction network (Figure 8). We used the MCC algorithm in cytoHubba plugin to screen the top 10 miRNA-genes in the network. Four miRNAs (hsa-miR-548am-3p, hsa-miR-513c-3p, hsa-miR-182-5p, and hsa-miR-548d-5p) and 6 genes (IGF1R, GSK3B, FOXO1, PRKAR2B, HIF1A, and PIK3R1) were the core nodes of the network (Table 2, Figure 9), and hsa-miR-548am-3p was considered as the core regulator because it targeted these 6 genes.
Discussion
In this study, mRNA and miRNA co-expression networks of hypertensive patients samples were constructed by using the WGCNA method. Out of the 19 identified mRNA modules, Msaddlebrown and Mgreenyellow had the most significant correlations to hypertension. For miRNAs, we identified 14 modules, of which Msalmon was the most significantly associated with hypertension. Finally, we identified 4 hub genes in the Msaddlebrown module, 8 hub genes in the Mgreenyellow module, and 7 hub miRNAs in the Msalmon module that were correlate with hypertension. Four miRNAs and 6 genes were also associated with the genetic susceptibility to hypertension. Our findings help us better understand the pathogenesis of hypertension, which in turn will provide us candidate biomarkers for clinical decision-making, potential therapeutic targets for accurate diagnosis, and treatment targets of hypertension.
In the Msaddlebrown module, the GO analysis indicates that Msaddlebrown mainly refered to membrane formation, translation, and ribosome and rRNA processing. For the Mgreenyellow module, the insulin signaling pathway and the HIF-1 signaling pathway were enriched. The HIF-1 signaling pathway is reported to be important for development of pulmonary hypertension in chronic hypoxia [23]. The HIF-1 pathway is related to proliferation of pulmonary arterial smooth muscle cells (PASMCs), which is also a central pathological component for a kind of hypertension [24]. We found 12 hub genes associated with hypertension (
Some hypertension-related pathways were found in the Msalmon module, which indicates that miRNA modules may also play a role in hypertension. The pathways include the adipocytokine signaling pathway, the thyroid hormone signaling pathway, the insulin signaling pathway, and the HIF-1 signaling pathway. The 3 key modules (Msaddlebrown, Mgreenyellow and Msalmon) have significant relations to hypertension, so we speculate that there may be regulatory relationships of the miRNAs and genes in the same pathway that affect blood pressure. In order to find out their relationship, we constructed a miRNA-gene regulatory network (Figure 8). We found 10 core nodes of the network (
This study had several limitations. First, our sample size was small and may not fully represent hypertension patients. Second, there is no biological experimental verification of the hub genes (miRNAs) and relationship between genes and miRNA. In a follow-up study, the molecular verification experiment will be conducted to uncover the molecular level mechanisms of miRNA-gene interactions and their relations to hypertension. Verified molecular markers can be used as new diagnostic indexes of hypertension in the future.
Conclusions
This study established a WGCNA-based gene expression data process workflow, identified 2 mRNA modules and 1 miRNA module related to hypertension, and provided potential candidate biomarkers for hypertension treatment. Our analysis revealed novel miRNA-gene interactions as well as central miRNAs and genes that play critical roles in hypertension.
Figures
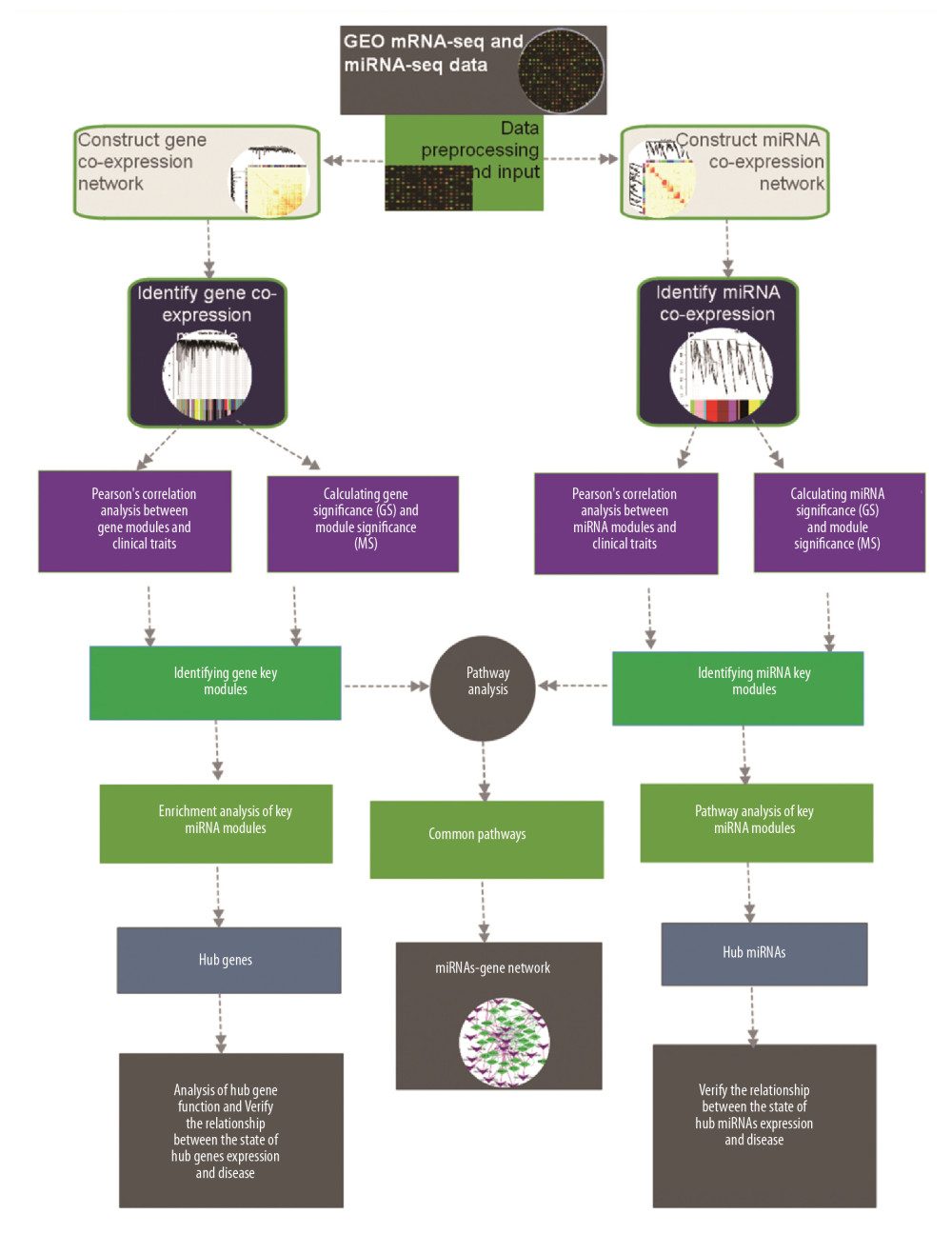 Figure 1. The workflow of this study.
Figure 1. The workflow of this study. 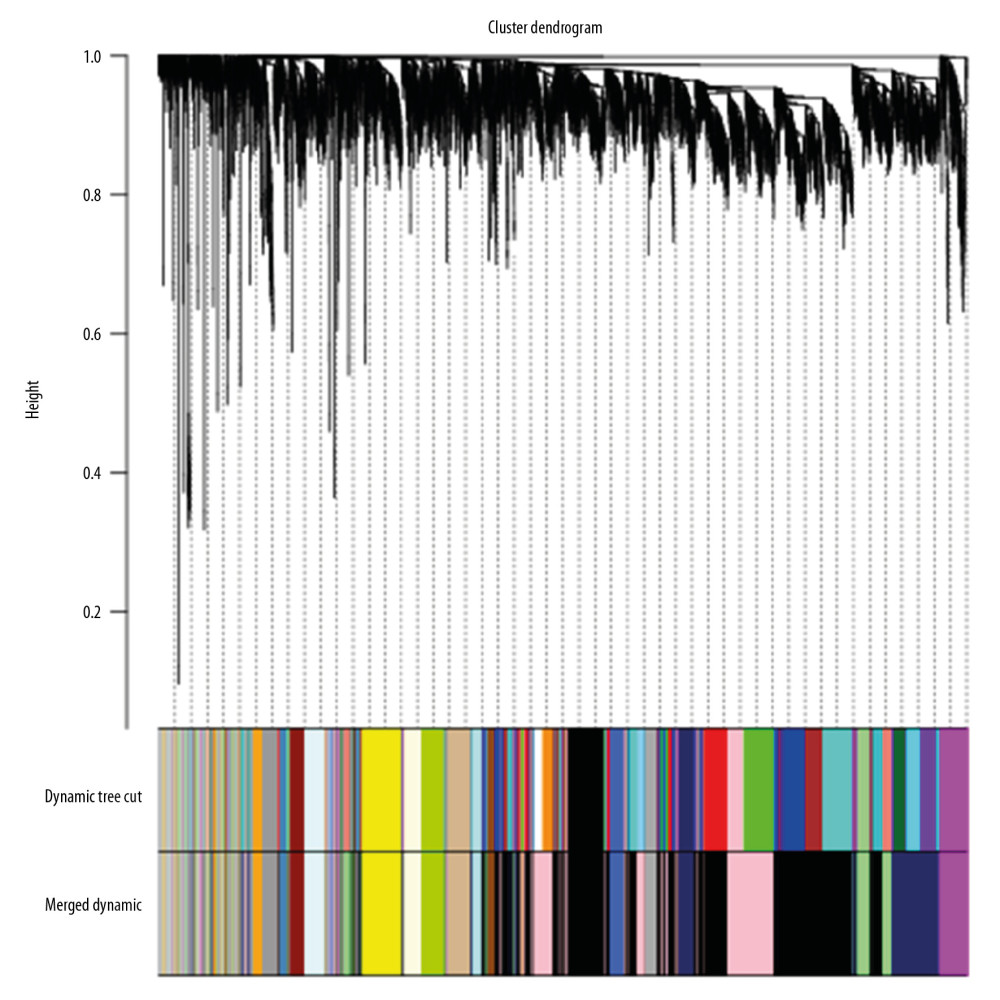 Figure 2. The cluster dendrogram of mRNA in mRNA expression data, each branch represents a gene, and each color below represents a co-expression module. The first ribbon represents the module detected by dynamic tree cutting, and the second ribbon represents the module after merging the similar module.
Figure 2. The cluster dendrogram of mRNA in mRNA expression data, each branch represents a gene, and each color below represents a co-expression module. The first ribbon represents the module detected by dynamic tree cutting, and the second ribbon represents the module after merging the similar module. 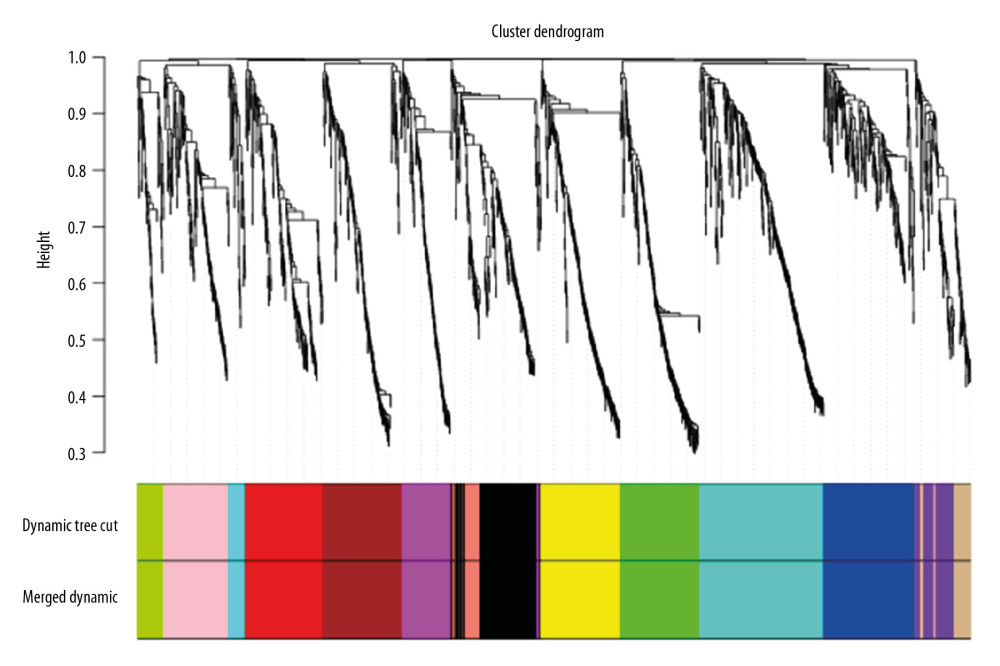 Figure 3. The cluster dendrogram of miRNAs in miRNA expression data.
Figure 3. The cluster dendrogram of miRNAs in miRNA expression data. 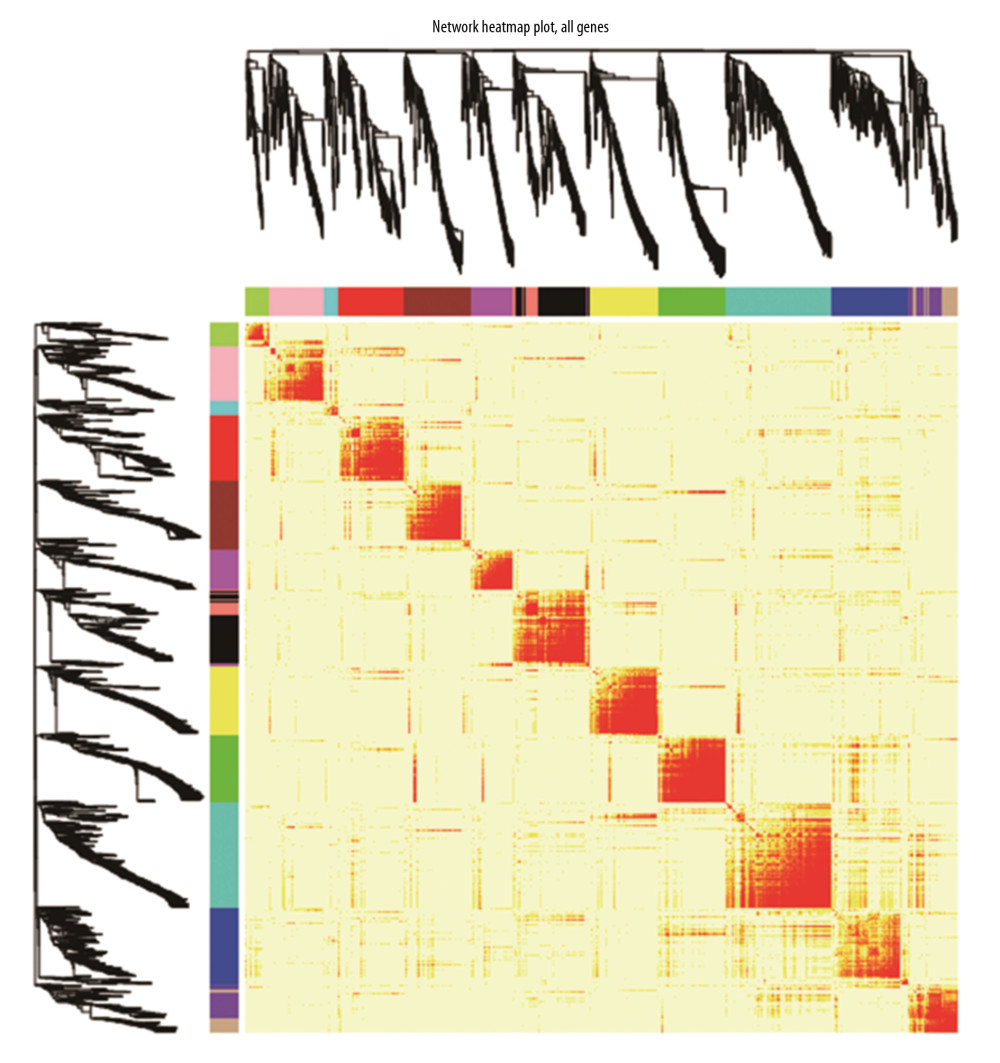 Figure 4. The interaction relationship of co-expressed miRNAs. The different colors in the vertical and horizontal axes stand for different miRNAs modules. The yellow color in the middle area indicated a degree of connection for each miRNA module.
Figure 4. The interaction relationship of co-expressed miRNAs. The different colors in the vertical and horizontal axes stand for different miRNAs modules. The yellow color in the middle area indicated a degree of connection for each miRNA module. 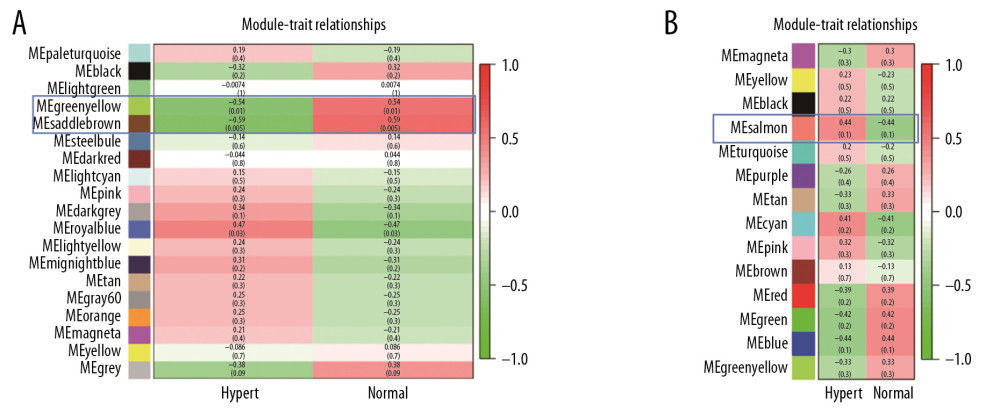 Figure 5. (A) mRNA module-trait relationship. The MESaddlebrown module was most significantly related to hypertension, and the MEGreenyellow module was the second one. (B) miRNA module-trait relationship. The MESalmon module was most significantly related to hypertension.
Figure 5. (A) mRNA module-trait relationship. The MESaddlebrown module was most significantly related to hypertension, and the MEGreenyellow module was the second one. (B) miRNA module-trait relationship. The MESalmon module was most significantly related to hypertension. 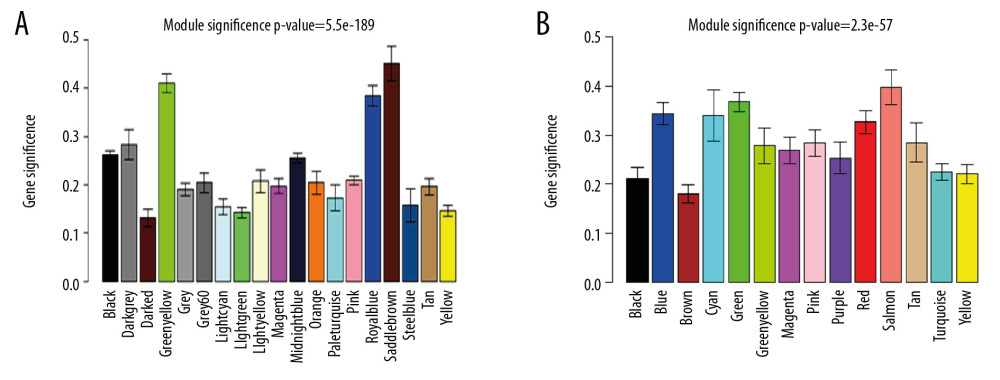 Figure 6. (A) Module significance of mRNA. Distribution of average mRNA significance and errors in the modules related to hypertension. Salmon module is associated with high blood pressure. (B) Module significance of mRNA.
Figure 6. (A) Module significance of mRNA. Distribution of average mRNA significance and errors in the modules related to hypertension. Salmon module is associated with high blood pressure. (B) Module significance of mRNA. 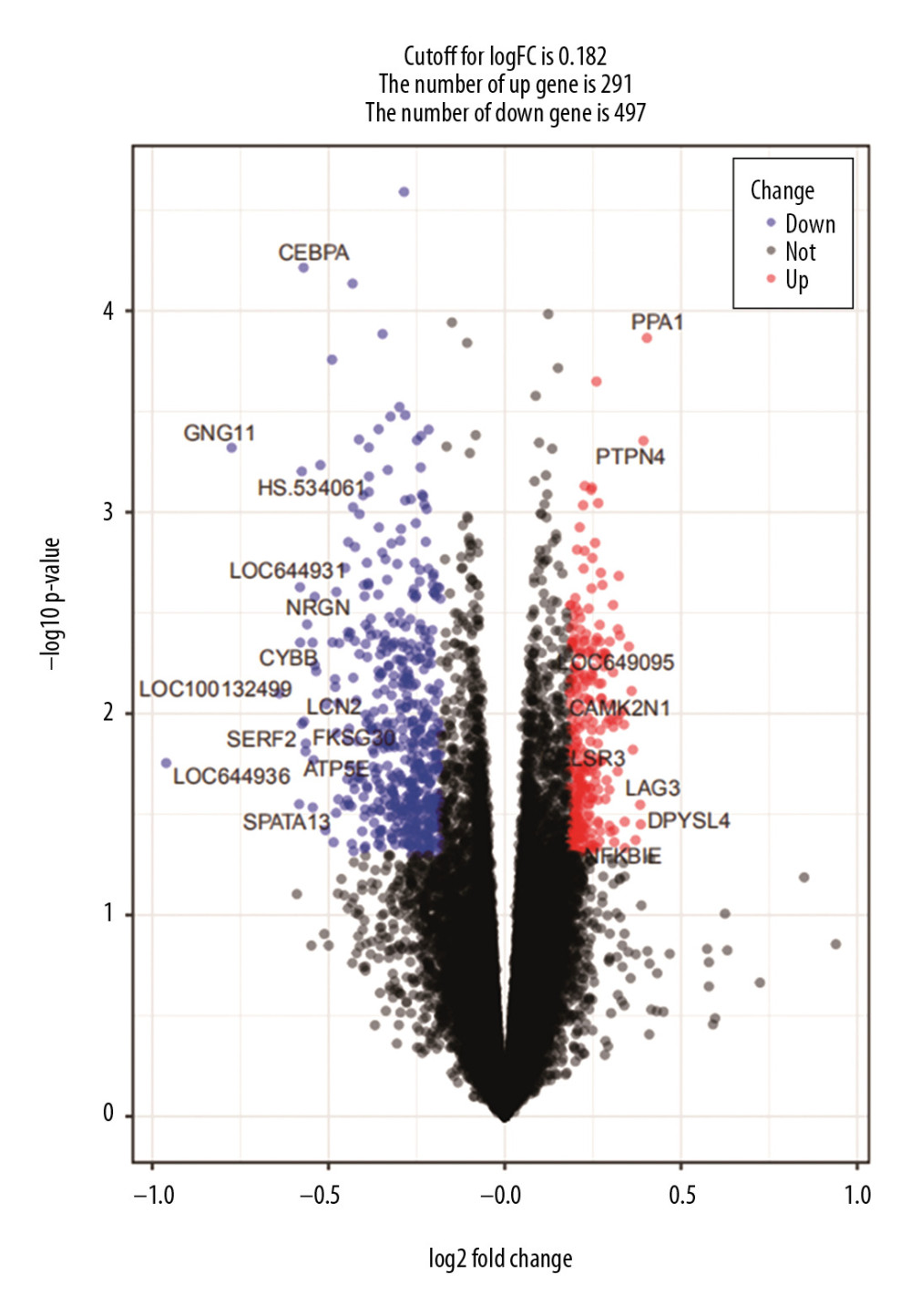 Figure 7. Volcano map of DEGs for GSE75360 dataset. The red dots on the right corresponds to 2-fold up changes with P-value less than 0.05, and the blue dots on the left means 2-fold down changes with P-value less than 0.05.
Figure 7. Volcano map of DEGs for GSE75360 dataset. The red dots on the right corresponds to 2-fold up changes with P-value less than 0.05, and the blue dots on the left means 2-fold down changes with P-value less than 0.05. 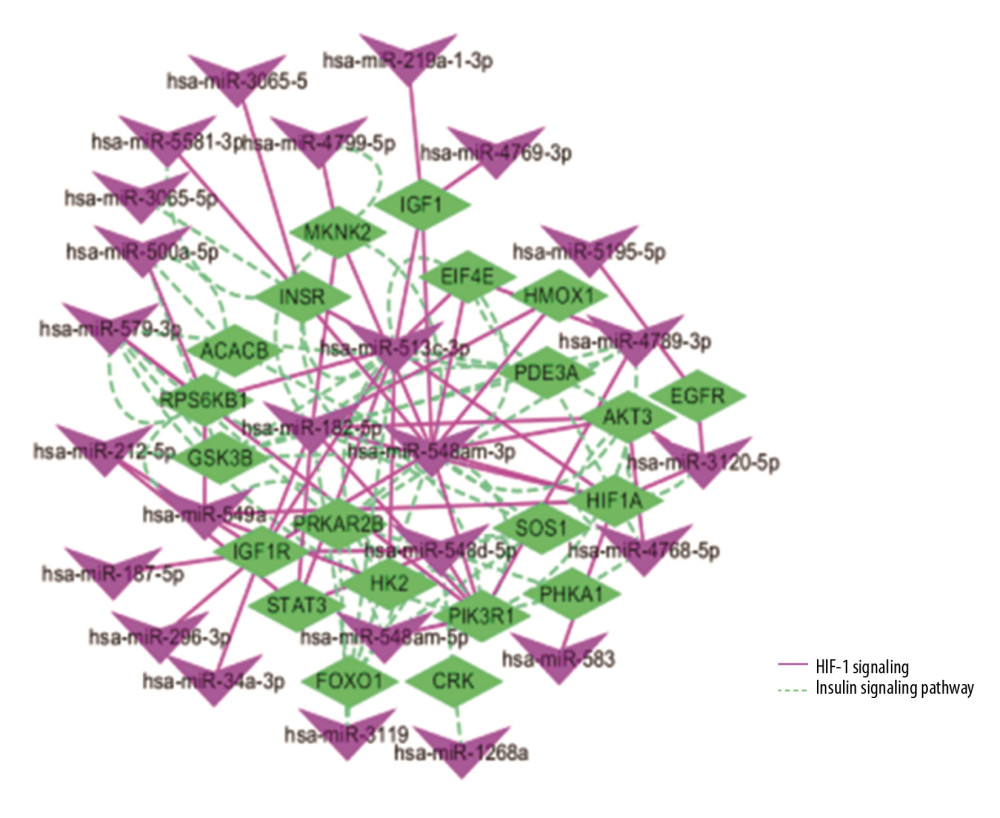 Figure 8. The regulatory network of miRNAs and target genes. Among them, the green prisms are mRNA, the purple triangles are miRNA, pink solid lines represent the HIF-1 pathway, and the green dotted lines represent the insulin pathway.
Figure 8. The regulatory network of miRNAs and target genes. Among them, the green prisms are mRNA, the purple triangles are miRNA, pink solid lines represent the HIF-1 pathway, and the green dotted lines represent the insulin pathway. 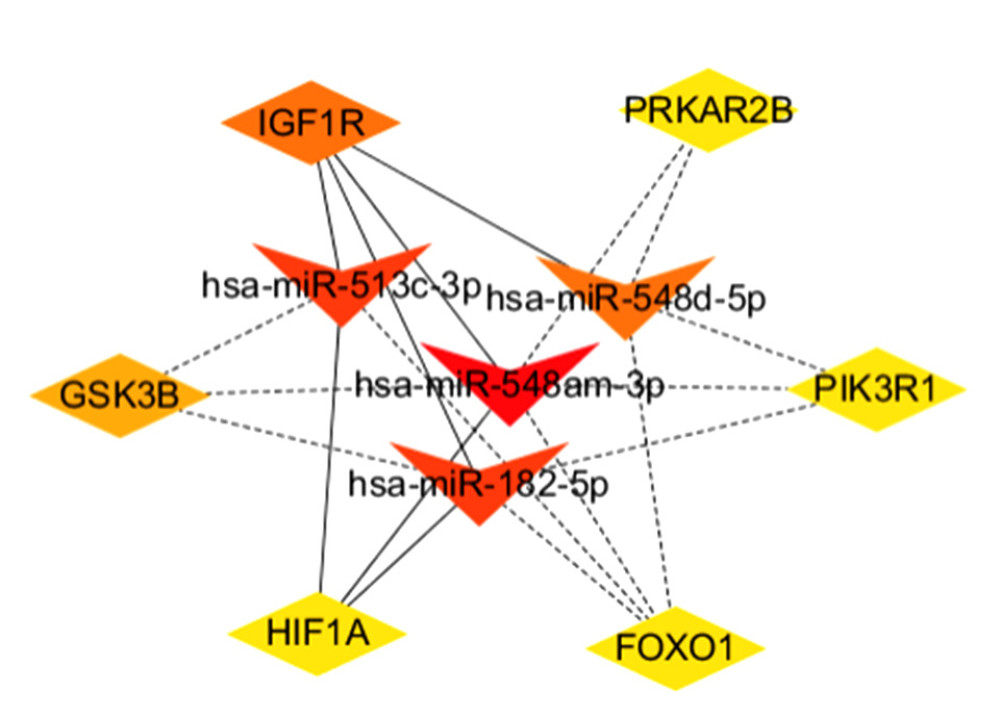 Figure 9. Top 10 nodes in miRNA-gene network.
Figure 9. Top 10 nodes in miRNA-gene network. Tables
Table 1. Hub genes (MiRNAs) in key modules.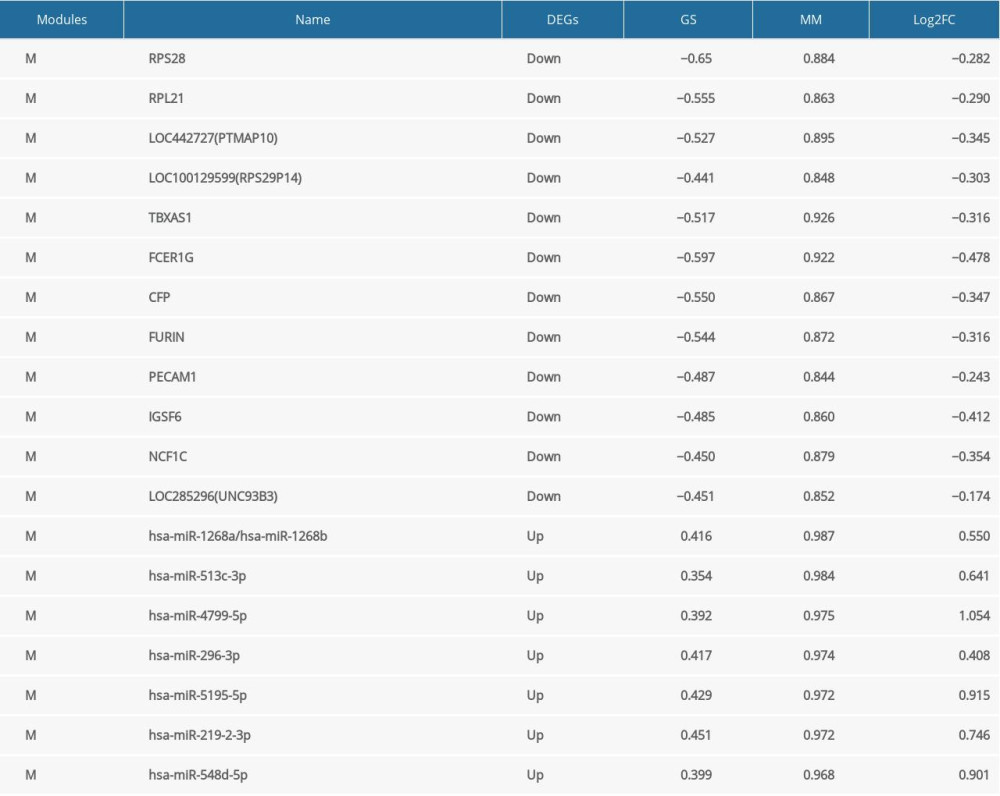 Table 2. Top 10 in network MiRNA-gene ranked by MCC method.
Table 2. Top 10 in network MiRNA-gene ranked by MCC method.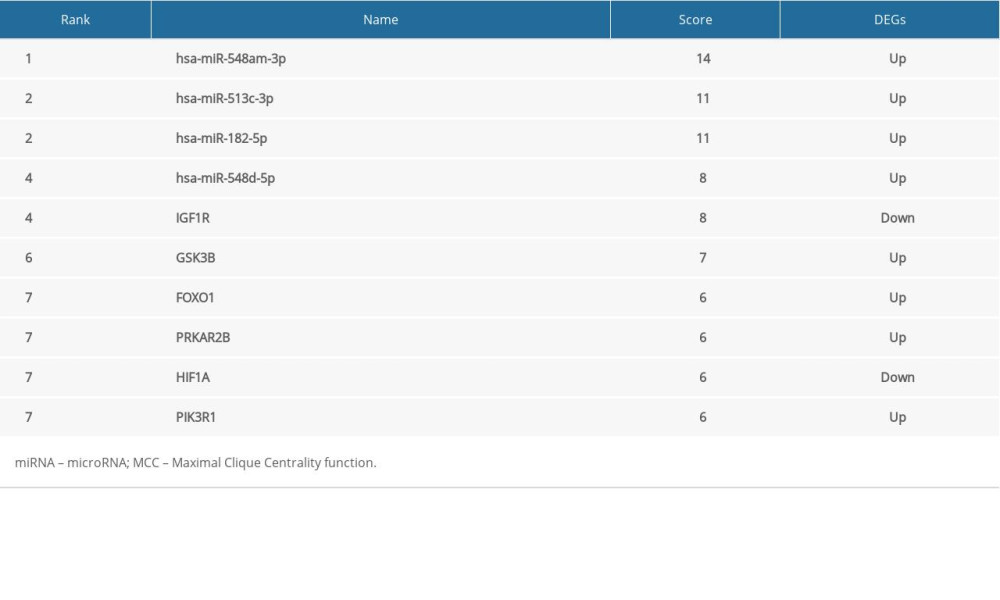 Supplementary Table 3. The results of salmon module analysis in mirPath.
Supplementary Table 3. The results of salmon module analysis in mirPath.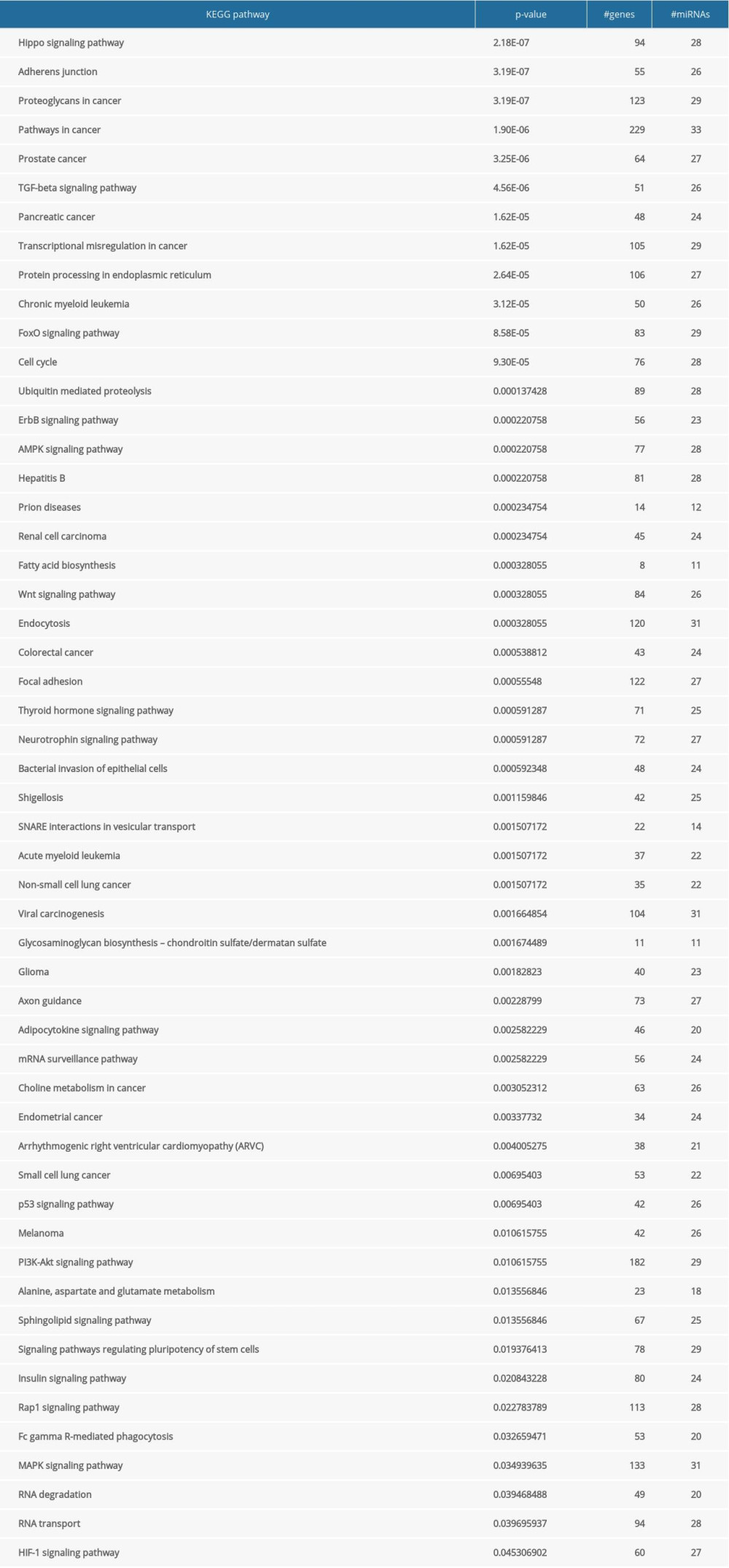 Supplementary Table 4. 26 hub genes identified in the saddlebrown.
Supplementary Table 4. 26 hub genes identified in the saddlebrown.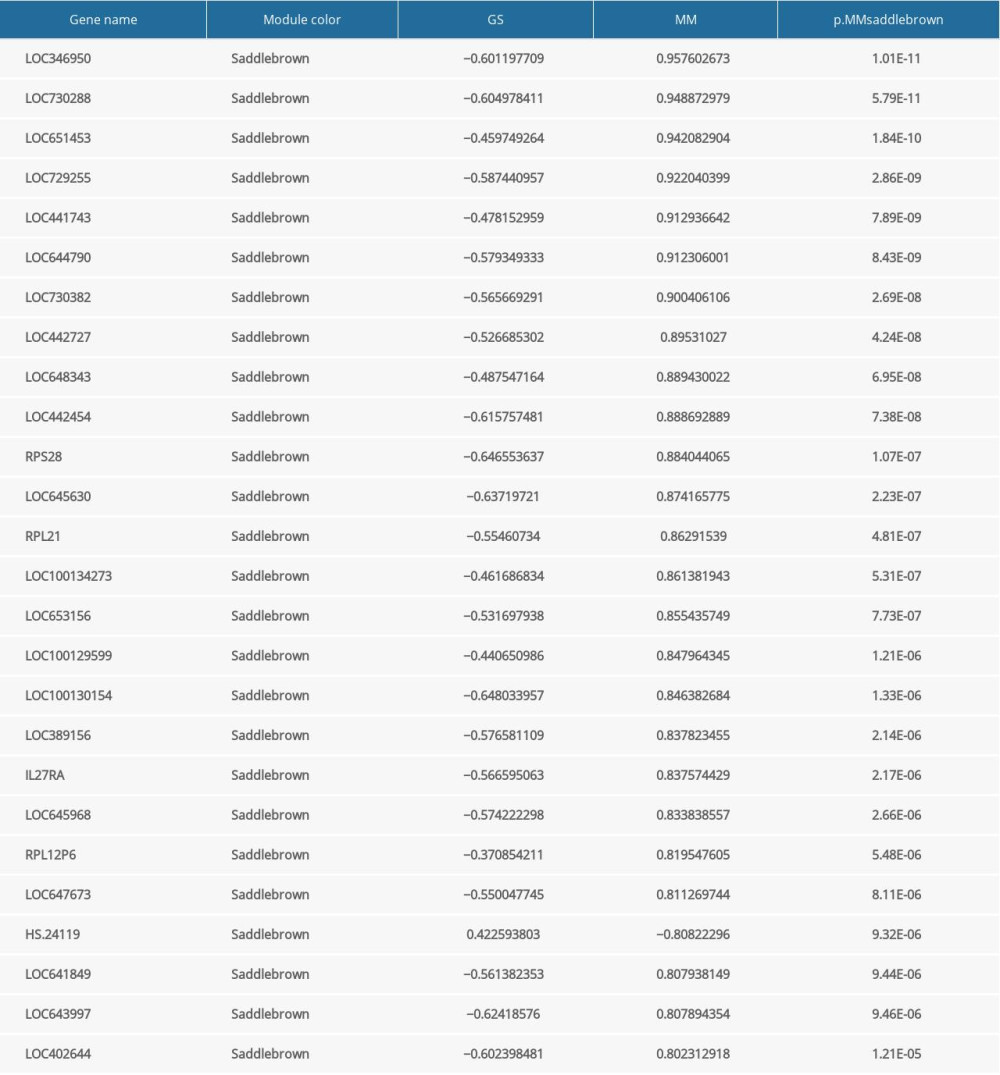 Supplementary Table 5. 53 hub genes identified in the greenyellow.
Supplementary Table 5. 53 hub genes identified in the greenyellow.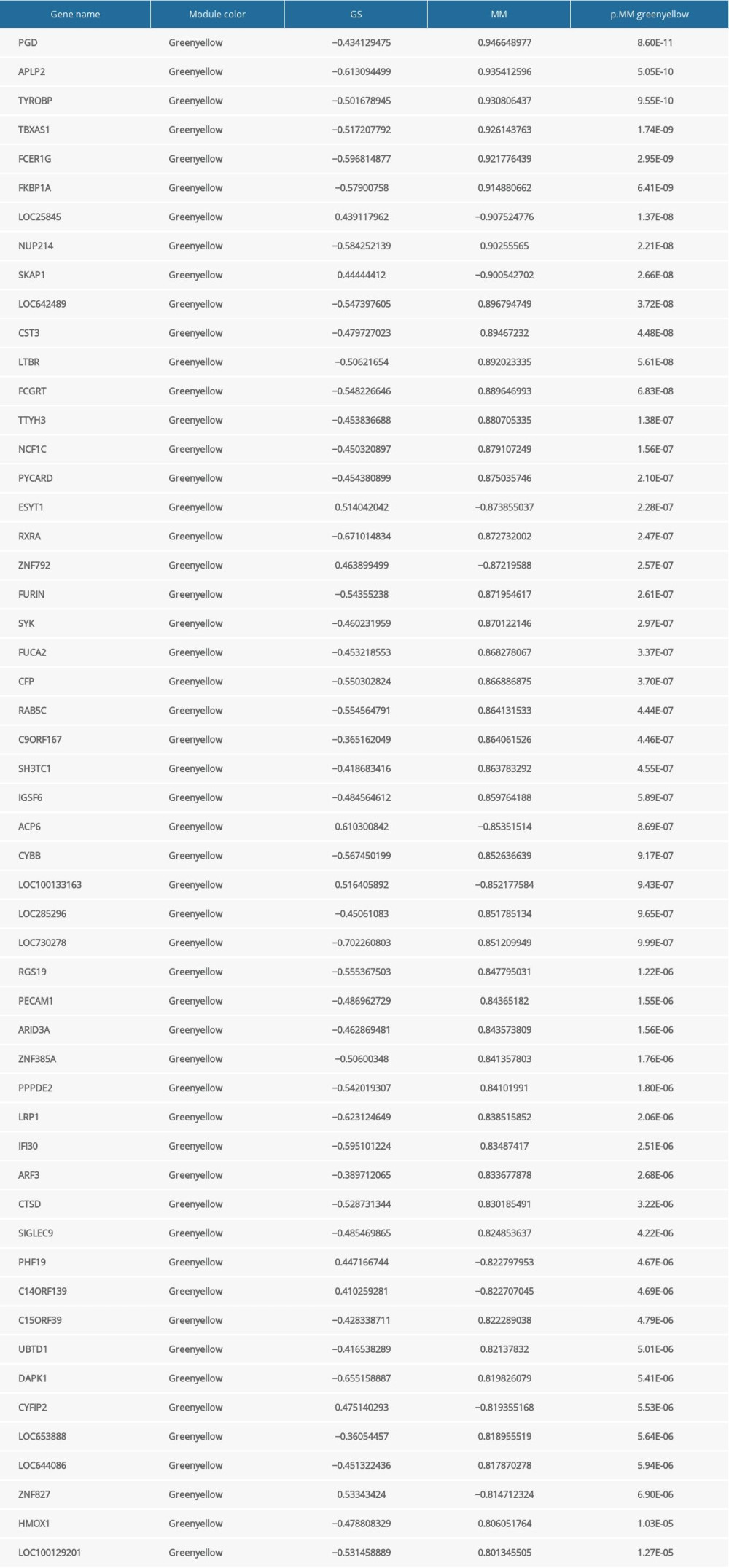 Supplementary Table 6. 22 hub genes identified in the salmon.
Supplementary Table 6. 22 hub genes identified in the salmon.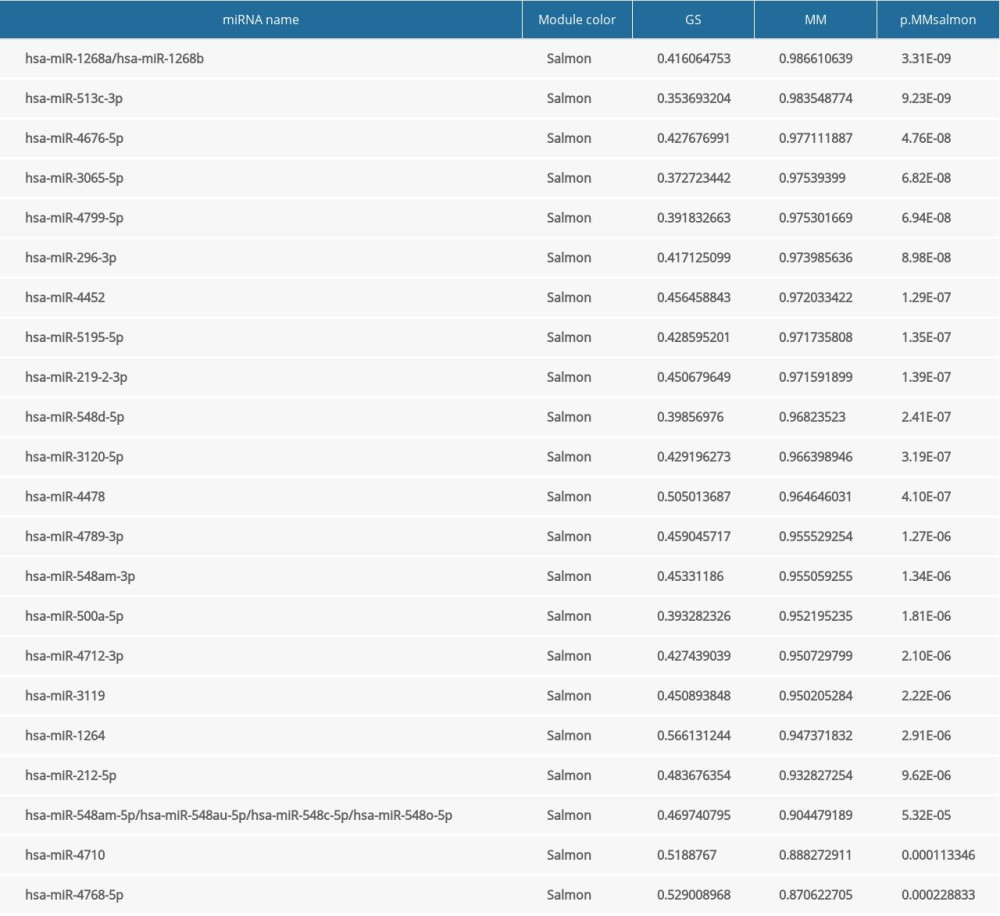
References
1. World Health Organization: A global brief on hypertension: Silent killer, global public health crisis: World Health Day, 2013 Available from: https://www.who.int/cardiovascular_diseases/publications/global_brief_hypertension/en/
2. Lewington S, Lacey B, Clarke R, The burden of hypertension and associated risk for cardiovascular mortality in China: JAMA Intern Med, 2016; 176(4); 524-32
3. Nonaka-Sarukawa M, Okada T, Ito T, Adeno-associated virus vector-mediated systemic interleukin-10 expression ameliorates hypertensive organ damage in Dahl salt-sensitive rats: J Gene Med, 2008; 10(4); 368-74
4. Wang X, Cade R, Sun Z, Human eNOS gene delivery attenuates cold-induced elevation of blood pressure in rats: Am J Physiol Heart Circ Physiol, 2005; 289(3); H1161-68
5. Hock FJ: Drug discovery and evaluation: Pharmacological Assays, 2016, Berlin, Heidleberg, Springer
6. Dluzen DF, Noren Hooten N, Zhang Y, Racial differences in microRNA and gene expression in hypertensive women: Sci Rep, 2016; 6; 35815
7. Wu X, Fan R, Identifications of potential therapeutic targets and drugs in angiotensin II-induced hypertension: Medicine (Baltimore), 2017; 96(46); e8501
8. Kontaraki JE, Marketou ME, Zacharis EA, MicroRNA-9 and microRNA-126 expression levels in patients with essential hypertension: Potential markers of target-organ damage: J Am Soc Hypertens, 2014; 8(6); 368-75
9. Jeong D, Kim J, Nam J, MicroRNA-124 links p53 to the NF-kappaB pathway in B-cell lymphomas: Leukemia, 2015; 29(9); 1868-74
10. Rhodes CJ, Wharton J, Boon RA, Reduced microRNA-150 is associated with poor survival in pulmonary arterial hypertension: Am J Respir Crit Care Med, 2013; 187(3); 294-302
11. Langfelder P, Horvath S, WGCNA: An R package for weighted correlation network analysis: BMC Bioinformatics, 2008; 9; 559
12. Liu X, Hu AX, Zhao JL, Identification of key gene modules in human osteosarcoma by co-expression analysis weighted gene co-expression network analysis (WGCNA): J Cell Biochem, 2017; 118(11); 3953-59
13. Zhang X, Feng H, Li Z, Application of weighted gene co-expression network analysis to identify key modules and hub genes in oral squamous cell carcinoma tumorigenesis: Onco Targets Ther, 2018; 11; 6001-21
14. Yuan L, Chen L, Qian K, Co-expression network analysis identified six hub genes in association with progression and prognosis in human clear cell renal cell carcinoma (ccRCC): Genom Data, 2017; 14; 132-40
15. Yin K, Zhang Y, Zhang S, Using weighted gene co-expression network analysis to identify key modules and hub genes in tongue squamous cell carcinoma: Medicine (Baltimore), 2019; 98(37); e17100
16. Liu J, Li S, Liang J, ITLNI identified by comprehensive bioinformatic analysis as a hub candidate biological target in human epithelial ovarian cancer: Cancer Manag Res, 2019; 11; 2379-92
17. Tang J, Kong D, Cui Q, Prognostic genes of breast cancer identified by gene co-expression network analysis: Front Oncol, 2018; 8; 374
18. Wang Q, Roy B, Dwivedi Y, Co-expression network modeling identifies key long non-coding RNA and mRNA modules in altering molecular phenotype to develop stress-induced depression in rats: Transl Psychiatry, 2019; 9(1); 125
19. Giulietti M, Occhipinti G, Principato G, Weighted gene co-expression network analysis reveals key genes involved in pancreatic ductal adenocarcinoma development: Cell Oncol (Dordr), 2016; 39(4); 379-88
20. Zhou XG, Huang XL, Liang SY, Identifying miRNA and gene modules of colon cancer associated with pathological stage by weighted gene co-expression network analysis: Onco Targets Ther, 2018; 11; 2815-30
21. Chin CH, Chen SH, Wu HH, cytoHubba: Identifying hub objects and sub-networks from complex interactome: BMC Syst Biol, 2014; 8(Suppl 4); S11
22. Bardou P, Mariette J, Escudie F, jvenn: An interactive Venn diagram viewer: BMC Bioinformatics, 2014; 15; 293
23. Bryant AJ, Carrick RP, McConaha ME, Endothelial HIF signaling regulates pulmonary fibrosis-associated pulmonary hypertension: Am J Physiol Lung Cell Mol Physiol, 2016; 310(3); L249-62
24. Xiao Y, Peng H, Hong C, PDGF promotes the Warburg effect in pulmonary arterial smooth muscle cells via activation of the PI3K/AKT/mTOR/HIF-1alpha signaling pathway: Cell Physiol Biochem, 2017; 42(4); 1603-13
25. Versari D, Daghini E, Virdis A, Endothelium-dependent contractions and endothelial dysfunction in human hypertension: Br J Pharmacol, 2009; 157(4); 527-3
26. Lopez-Parra V, Malaria B, Lopez-Franco O, Fact receptor deficiency attenuates diabetic nephropathy: J Am Soc Nephrol, 2012; 23(9); 1518-27
27. Li N, Luo W, Jurong Z, Associations between genetic variations in the FURIN gene and hypertension: BMC Med Genet, 2010; 11; 124
28. Zeisel H, Livingston JC, Schettino C, Serum levels of adhesion molecules in women with pregnancy-induced hypertension: Wien Klin Wochenschr, 2001; 113(15–16); 588-92
29. Wang WC, Chiu YF, Chung RH, IGF1 gene is associated with triglyceride levels in subjects with family history of hypertension from the SAPPHIRe and TWB projects: Int J Med Sci, 2018; 15(10); 1035-42
30. Sun M, Ramchandran R, Chen J, Smooth muscle insulin-like growth factor-1 mediates hypoxia-induced pulmonary hypertension in neonatal mice: Am J Respir Cell Mol Biol, 2016; 55(6); 779-91
31. Takahashi J, Orcholski M, Yuan K, PDGF-dependent β-catenin activation is associated with abnormal pulmonary artery smooth muscle cell proliferation in pulmonary arterial hypertension: FEBS Lett, 2016; 590(1); 101-9
32. Qi Y, Zhang K, Wu Y, Novel mechanism of blood pressure regulation by forkhead box class O1-mediated transcriptional control of hepatic angiotensinogen: Hypertension (Dallas, Tex: 1979), 2014; 64(5); 1131-40
33. Kinoshita K, Ashenagar MS, Tabuchi M, Whole rat DNA array survey for candidate genes related to hypertension in kidneys from three spontaneously hypertensive rat substrains at two stages of age and with hypotensive induction caused by hydralazine hydrochloride: Exp Ther Med, 2011; 2(2); 201-12
34. Sheng ZL, Ju CW, Yan GL, The relevance of HIF1A gene polymorphisms and primary hypertensive left ventricular hypertrophy in Chinese Han population: Eur Rev Med Pharmacol Sci, 2019; 23(18); 8095-100
35. Wang Y, Huang X, Leng D, DNA methylation signatures of pulmonary arterial smooth muscle cells in chronic thromboembolic pulmonary hypertension: Physiol Genom, 2018; 50(5); 313-22
Figures
Figure 1. The workflow of this study.Figure 2. The cluster dendrogram of mRNA in mRNA expression data, each branch represents a gene, and each color below represents a co-expression module. The first ribbon represents the module detected by dynamic tree cutting, and the second ribbon represents the module after merging the similar module.Figure 3. The cluster dendrogram of miRNAs in miRNA expression data.Figure 4. The interaction relationship of co-expressed miRNAs. The different colors in the vertical and horizontal axes stand for different miRNAs modules. The yellow color in the middle area indicated a degree of connection for each miRNA module.Figure 5. (A) mRNA module-trait relationship. The MESaddlebrown module was most significantly related to hypertension, and the MEGreenyellow module was the second one. (B) miRNA module-trait relationship. The MESalmon module was most significantly related to hypertension.Figure 6. (A) Module significance of mRNA. Distribution of average mRNA significance and errors in the modules related to hypertension. Salmon module is associated with high blood pressure. (B) Module significance of mRNA.Figure 7. Volcano map of DEGs for GSE75360 dataset. The red dots on the right corresponds to 2-fold up changes with P-value less than 0.05, and the blue dots on the left means 2-fold down changes with P-value less than 0.05.Figure 8. The regulatory network of miRNAs and target genes. Among them, the green prisms are mRNA, the purple triangles are miRNA, pink solid lines represent the HIF-1 pathway, and the green dotted lines represent the insulin pathway.Figure 9. Top 10 nodes in miRNA-gene network. Tables
Table 1. Hub genes (MiRNAs) in key modules.Table 2. Top 10 in network MiRNA-gene ranked by MCC method.Table 1. Hub genes (MiRNAs) in key modules.Table 2. Top 10 in network MiRNA-gene ranked by MCC method.Supplementary Table 3. The results of salmon module analysis in mirPath.Supplementary Table 4. 26 hub genes identified in the saddlebrown.Supplementary Table 5. 53 hub genes identified in the greenyellow.Supplementary Table 6. 22 hub genes identified in the salmon. In Press
Clinical Research
Body Weight and Insulin Resistance Indicators Among ChildrenMed Sci Monit In Press; DOI: 10.12659/MSM.951434
Clinical Research
Comparison of Radiographic Cervical Sagittal Alignment Parameters in Patients With Nonspecific Neck Pain, D...Med Sci Monit In Press; DOI: 10.12659/MSM.952950
Clinical Research
Combined Fibrinogen and Urinary α1-Microglobulin as Predictors of Respiratory Tract Infection in Children w...Med Sci Monit In Press; DOI: 10.12659/MSM.951066
Database Analysis
Evaluation of Salivary Total Oxidant Status (TOS) and Total Antioxidant Status (TAS) in Orthodontic Patient...Med Sci Monit In Press; DOI: 10.12659/MSM.952052
Most Viewed Current Articles
17 Jan 2024 : Review article 14,175,576
Vaccination Guidelines for Pregnant Women: Addressing COVID-19 and the Omicron VariantDOI :10.12659/MSM.942799
Med Sci Monit 2024; 30:e942799
13 Nov 2021 : Clinical Research 3,756,620
Acceptance of COVID-19 Vaccination and Its Associated Factors Among Cancer Patients Attending the Oncology ...DOI :10.12659/MSM.932788
Med Sci Monit 2021; 27:e932788
14 Dec 2022 : Clinical Research 2,465,966
Prevalence and Variability of Allergen-Specific Immunoglobulin E in Patients with Elevated Tryptase LevelsDOI :10.12659/MSM.937990
Med Sci Monit 2022; 28:e937990
16 May 2023 : Clinical Research 708,651
Electrophysiological Testing for an Auditory Processing Disorder and Reading Performance in 54 School Stude...DOI :10.12659/MSM.940387
Med Sci Monit 2023; 29:e940387