02 August 2020: Database Analysis
Prognostic Implications of Novel Gene Signatures in Gastric Cancer Microenvironment
Mengyu Sun1ACE, Jieping Qiu1BCF, Huazheng Zhai1BDF, Yaoqun Wang1DF, Panpan Ma1EF, Mengyin Li1E, Bo Chen2AG*DOI: 10.12659/MSM.924604
Med Sci Monit 2020; 26:e924604






Abstract
BACKGROUND: Increasing studies have shown the important clinical role of immune and stromal cells in gastric cancer microenvironment. Based on information of immune and stromal cells in The Cancer Genome Atlas, this study aimed to construct a prognostic risk assessment model for gastric cancer.
MATERIAL AND METHODS: Based on the immune/structural scores, differentially expressed genes (DEGs) were filtered and analyzed. Afterwards, DEGs associated with prognosis were screened and the risk assessment model was constructed in the training set. Moreover, the validity of the model was verified both in the testing set and the overall sample.
RESULTS: In this study, patients were divided into high-score and low-score groups based on immune/stromal score, and 919 DEGs were identified. By applying least absolute shrinkage and selection operator (LASSO) and Cox analysis, 10 mRNAs were selected to form a prognostic risk assessment model, risk score=(0.294*SLC17A9) + (–0.477*FERMT3) + (0.866*NRP1) + (0.350*MMRN1) + (0.381*RNASE1) + (0.189*TRIB3) + (0.230*PGAP3) + (0.087*MAGEA3) + (0.182*TACR2) + (0.368*CYP51A1). In the training set, the low-risk group divided by the model was found to have better overall survival, and the prediction efficiency of the model was demonstrated to be good. Multivariate Cox analysis indicated that the model could work as a prognostic factor independently. Similar results were shown in the testing group and overall patients cohort group. Finally, the risk assessment model and other clinical variables were integrated to construct a nomogram.
CONCLUSIONS: In general, this study constructs a prognostic risk assessment model for gastric cancer, which could improve the prognosis stratification of patients combined with other clinical indicators.
Keywords: Biological Markers, Computational Biology, Area Under Curve, Biomarkers, Tumor, carcinogenesis, Databases, Genetic, Gene Regulatory Networks, Immunity, Innate, Models, Genetic, Neoplasm Proteins, RNA, Messenger, RNA, Neoplasm, ROC Curve, Stromal Cells, tumor microenvironment
Background
Gastric cancer globally ranks as a frequent malignant cancer with high mortality, and the distribution of its prognosis outcomes is quite different. The 5-year overall survival (OS) rate for gastric cancer patients ranges from 10% with stage IV to over 80% with stage I in China [1–3]. The American Joint Committee on Cancer (AJCC) stage is currently the most commonly used tool for prognostic evaluation of gastric cancer [4–6]. With the continuous updating of the stage system, its predictive performance has also continuously improved [7–11]. Nevertheless, the prognosis of many patients with the same AJCC stage and similar clinical treatment methods still varies widely [12,13], indicating that the information provided by the stage for the prognosis assessment of gastric cancer is insufficient. Therefore, based on the existing stage system, new analytical strategies are needed to improve the effectiveness of gastric cancer prognosis assessment so that clinicians can make better clinical decisions in a timely manner.
Tumor microenvironment (TME) contains cellular and noncellular components. Cell components include tumor cells, inflammatory cells, immune cells, endothelial cells, mesenchymal stem cells, and tumor related fibroblasts, etc., and noncell components mainly include cytokines, chemokines, etc. [14,15]. According to the TME theory, the occurrence, progression, and prognosis of tumors are not only determined by the genetic and epigenetic factors of cancer cells, but also by the interaction between tumor cells and the surrounding milieu [16,17]. The various components of TME play a supporting role for tumor growth together [18,19]. Among them, immune and stromal cells are the main cell components, except tumor cells, which have been reported to have unique value in tumor diagnosis and prognosis evaluation in many studies [20–24]. Thus, combining the information contained in immune/stromal cells with common tumor clinical indicators such as AJCC stage to evaluate the prognosis of patients is conducive to optimize the prognosis stratification for gastric cancer patients.
ESTIMATE (Estimation of STromal and Immune cells in MAlignant Tumor tissues using Expression data) designed by Yoshihara et al. is available to predict the purity of the tumor as well as the degree of infiltration of non-tumor components [25]. Based on particular gene markers of immune/stromal cells, the algorithm calculates ESTIMATE/immune/stromal scores using gene expression information from The Cancer Genome Atlas (TCGA). The synthesis of immune and stromal score is the ESTIMATE score, and these 3 scores (immune scores, stromal scores, and ESTIMATE score) can be used to predict infiltration degree of immune/stromal cells. According to immune/stromal scores of gastric adenocarcinoma (STAD) in the algorithm, this current study identified differentially expressed genes (DEGs) in TME and performed least absolute shrinkage and selection operator (LASSO) and Cox analysis on DEGs to establish a prognostic risk assessment model.
Material and Methods
RAW DATA:
Level 3 gene expression profile of stomach adenocarcinoma patients in TCGA database was downloaded through the UCSC (
SCORES AND CLINICOPATHOLOGICAL INFORMATION:
Correlation between ESTIMATE/immune/stromal scores of 415 STAD patients and the clinicopathological characteristics containing stage and grade was analyzed. Statistical differences were measured by one-way ANOVA test, and
IDENTIFICATION OF DEGS:
Excluding samples with missing gene expression data, a total of 379 cases were available to screen for DEGs. Package limma of the R programming language was applied to identify DEGs according to immune and stromal scores individually. Fold change >1.5 and adjusted
FUNCTIONAL ANALYSIS OF DEGS IN STAD:
To annotate and analyze the biological functions of DEGs, the Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis and Gene Ontology (GO) analysis were carried out by R program. GO enrichment analysis is composed of 3 parts, namely MF (molecular function), BP (biological processes), and CC (cellular components). Subsequently, the online database STRING (
RANDOMIZED GROUPING:
Patients with survival time missing or less than 30 days were excluded in order to rule out the patient’s death from other factors rather than the consequence of the tumor. Ultimately, samples with missing gene expression and unqualified survival time were excluded; from the total 415 cases, 348 STAD patients were included in our study. Then the patients were split into 2 groups randomly: the training group and the testing group. Each group included 174 patients. The training group was utilized to learn sample features and estimate models, while the testing group was set as an internal verification queue to verify the prediction performance of the model.
STATISTICAL ANALYSIS AND CONSTRUCTION OF RISK ASSESSMENT MODEL:
Whether there was a difference in clinicopathological variables between the training group and the testing group was tested.
Afterwards, the LASSO and Cox regression model was used to filter DEGs which correlated with the prognosis of STAD patients. Through LASSO analysis, the minimum lambda (λ) value was selected as the optimal lambda parameter. Then a survival-related linear risk assessment model was constructed, the formula:
Next, 8 indicators (age, gender, grade, stage, stage_T, stage_N, stage_M and risk) were used to construct a nomogram to personally predict the patient’s 1-year, 3-year, and 5-year survival rate.
Statistical analyses and graphic drawing were accomplished using SPSS 23.0, GraphPad 7.0 and R program (3.6).
GENE SET ENRICHMENT ANALYSIS (GSEA):
Enrichment analyses of GO and KEGG were performed by GSEA v3.0 for all genes between the high-risk group and the low-risk group. Enrichment pathways were ranked based on normalized enrichment score (NSE).
Results
THE CORRELATION OF IMMUNE AND STROMAL SCORE WITH CLINICOPATHOLOGICAL INDICATORS:
Gene expression profiles and phenotype information of STAD patients were obtained and integrated from TCGA cohort. On the basis of ESTIMATE algorithm, immune scores ranged from −1568.74 to 2826.73, and stromal scores were distributed between −1957.19 to 2085.81. Estimate score was significantly related to grade and stage (Figure 1A, P<0.0001, Figure 1B, P=0.0023). In general, the higher the ESTIMATE score was related to the more progressive stage. As shown in Figure 1A and 1B, G3 had a higher ESTIMATE score than G2, and the ESTIMATE score of stage II, III, and IV was individually higher than stage I. The immune score was associated with grade significantly (Figure 1C, P<0.0001), while it showed no correlation with AJCC stage (Figure 1D, P=0.0625). The stromal score had significant relevance with grade (Figure 1E, P<0.0001) and AJCC stage (Figure 1F, P=0.0004).
IDENTIFICATION OF DEGS BASED ON IMMUNE/STROMAL SCORE:
To detect the relationship between gene expression and immune/stromal score, microarray data of STAD cases were compared. According to immune score, 1051 genes were high-expressed and 236 genes were down-expressed in the high score group compared with the low score group (Figure 2A). Similarly, 1591 high-expressed genes and 427 down-expressed genes were obtained based on stromal score (Figure 2B). In total, 769 DEGs were synchronously high-expressed in both high score groups (Figure 2C), while 150 genes were commonly down-expressed (Figure 2D). Heat map including 919 DEGs showed distinct gene expression profiles (Figure 2E).
FUNCTIONAL ENRICHMENT ANALYSIS AND ESTABLISHMENT OF PPI NETWORK:
Functional analysis containing GO and KEGG was applied in total 919 DEGs (Figure 3A, 3B). Thirty function annotations of each part were listed and the part of GO containing BP, CC, and MF. For the BP, leukocyte migration (GO: 0050900) and T cell activation (GO: 0042110) were the top 2 pathways. For the CC, DEGs were significantly enriched in collagen-containing extracellular matrix (GO: 0062023) and external side of plasma membrane (GO: 0009897). For MF, the genes were mostly involved in glycosaminoglycan binding (GO: 0005539) and extracellular matrix structural constituent (GO: 0005201). As regard to KEGG analysis, the significant enriched pathways were cytokine-cytokine receptor interaction (hsa04060) and chemokine signaling pathway (hsa04062).
PPI network was established based on 919 DEGs. Afterwards, a plug-in of Cytoscape named MCODE was used to identify the most important co-regulated modular. The top 2 significant modules were selected and are shown in Figure 3C and 3D. For the sake of convenience, we named these modules the C3 and TIMP1 modules, respectively. In the C3 module, 666 edges involving 37 nodes were formed in the network, C3, GNG2, GNB4, GNG11, C3AR1, and CXCL12 were the remarkable nodes, as they had more connection with other genes of the module. In the TIMP1 module which had 17 nodes with 136 degrees, TIMP1, APOA1, LTBP1, and FBN1 had higher degree values.
CONSTRUCTION OF RISK ASSESSMENT MODEL IN TRAINING GROUP:
There were 348 STAD cases included in the analysis that were allocated into a training set and a testing set randomly. And there was no statistically significant difference in clinical variables between the 2 groups (Supplementary Table 1). It could be considered that the 2 groups of cases originated from the same population, and the randomization was reasonable.
In order to identify the genes related to the prognosis of STAD patients in the training set, we used LASSO and Cox regression model to perform screening of variables on high-dimensional mRNA expression profile data. First, survival analysis of 919 DEGs was conducted in the training set. After 100 times of 10-fold cross-validation, 19 survival related mRNAs were screened out when λ was taken as the minimum value of 0.036 (Figure 4A, 4B). After optimization by Cox analysis, 10 mRNAs were chosen to establish the risk assessment model as shown in forest graph (Figure 4C). Extracting the coefficients of multivariate Cox analysis of mRNAs from the LASSO and Cox regression model, risk score=(0.294*SLC17A9)+ (−0.477*FERMT3)+(0.866*NRP1) +(0.350*MMRN1)+ (0.381*RNASE1)+(0.189*TRIB3)+(0.230*PGAP3)+(0.087*MAGEA3)+ (0.182*TACR2) + (0.368*CYP51A1).
Based on the aforementioned formula, risk value was calculated for every patient in the training set. The median risk value of 0.97 was used as the cutoff value, and 174 cases in training set were allocated into high-risk group (>0.97, n=87) and low-risk group (<0.97, n=87) (Figure 5A). In the high-risk group, the average survival time of patients was shorter and the number of deaths was higher (Figure 5B). Meanwhile, the heat map showed that the expression of 10 mRNAs in the 2 groups was also different (Figure 5C). To assess the predictive performance of risk assessment model, the ROC curve was plotted. And AUC (area under curve) of the 5-year survival rate of STAD patients was 0.815 (Figure 5D), indicating a good prediction efficiency of the model. As shown in the Kaplan-Meier survival curve, OS of patients in high risk group was shorter than that in low risk group (P<0.001) (Figure 5E).
VALIDATION OF RISK ASSESSMENT MODEL IN TESTING SET AND TOTAL PATIENTS:
In order to further verify the risk assessment model, the risk value of every patient in testing set was calculated according to the risk calculation formula. After that, cases were allocated into high and low risk groups based on the risk cut-off value (0.97) of the training set.
In testing set, patients had shorter survival time and more deaths in the high-risk group (Figure 6A, 6B). Meanwhile, the heat map displayed that there was also difference in the expression of 10 mRNAs between the 2 groups (Figure 6C). The AUC value of the 5-year survival rate for patients in the testing set was 0.781 (Figure 6D), indicating that the model still had good prediction effect. The survival analysis demonstrated that the OS of patients was shorter in high-risk group (P<0.001) (Figure 6E).
The training set and the testing set were combined into an overall patients cohort. As depicted in Figure 7A–7C, the average survival time of cases was shorter and the number of deaths was larger in high-risk group, while the expression of 10 mRNAs in the high-risk group and the low-risk group was different. In the overall STAD patients cohort, the prediction effect of the model was also favorable, the AUC value of the 5-year survival rate was 0.789 (Figure 7D). And the high-risk group showed a worse OS (P<0.001) (Figure 7E).
COX REGRESSION ANALYSIS AND CONSTRUCTION OF NOMOGRAM:
To assess whether the risk assessment model was an independent factor affecting the prognosis of STAD, univariate and multivariate Cox regression analysis was applied to the training set, testing set and overall patients cohort group, respectively. The included clinical variables contained age, gender, grade, stage_T, stage_N, stage_M, stage, and risk model. In the univariate Cox regression analysis, poor OS of the overall patients cohort were significantly correlated with age (ref. ≤65), grade (ref. 1–2), stage_T (ref. T1–T2), stage_N (ref. N0), stage (ref. I–II) and risk model (ref. low). After adjustment of multivariate Cox analysis, the risk assessment model was still remarkably related to the OS of patients in the 3 cohorts, indicating that the model can work as a prognostic index independently. The detailed information is shown in Table 1.
By integrating multiple risk factors, the nomogram can be an effective tool to quantitatively assess an individual’s risk in a clinical setting. On the basis of the aforementioned clinical variables, a nomogram was established to forecast the probability of 1-year, 3-year and 5-year OS (Figure 8).
GSEA IN GO AND KEGG GENE SETS:
GSEA analysis was performed to unveil the pathways that genes significantly enriched in GO and KEGG gene sets. In the GO gene set (Figure 9A), the high-risk group was remarkably correlated with neuron projection cytoplasm (NSE=2.306, P=0.000), intercalated disc (NSE=2.305, P=0.000), negative regulation of protein polymerization (NSE=2.280, P=0.000), and cell cell contact zone (NSE=2.268, P=0.000), while the low-risk group was negatively related to translational initiation (NSE=−2.140, P=0.004), RNA phosphodiester bond hydrolysis (NSE=−2.095, P=0.000), translation initiation factor activity (NSE=−2.090, P=0.000), and nuclear transcribed mRNA catabolic process (NSE=−2.084, P=0.008). For the KEGG gene set (Figure 9B), the high-risk group was significantly associated with gap junction (NSE=2.222, P=0.000), vascular smooth muscle contraction (NSE=2.103, P=0.000), melanogenesis (NSE=2.095, P=0.000), and focal adhesion (NSE=2.079, P=0.000). And the low-risk group was negatively correlated with RNA degradation (NSE=−1.984, P=0.002), primary immunodeficiency (NSE=−1.939, P=0.000), aminoacyl tRNA biosynthesis (NSE=−1.922, P=0.011), and proteasome (NSE=−1.918, P=0.015).
Discussion
With the development of tumor microenvironment (TME) theory, in addition to the intrinsic characteristics of tumor cells, the role of external microenvironment in tumor progression has attracted increasing attention. Through interaction with tumor cells and immune/stromal cells produce an effect in the stages of tumor occurrence, progression, invasion, metastasis, recurrence, and drug response, so as to affect the prognosis of patients [21,22]. Seeing that the ideal outcome of patients’ prognosis assessment cannot be obtained by relying mainly on AJCC stage, analysis of TME can provide supplementary information for prognostic risk evaluation of patients, thereby improving the effectiveness of risk assessment.
Based on the immune score and stromal score of gastric adenocarcinomas, this study screened 919 differentially expressed genes by comparing the high-score group with the low-score group. Subsequently, functional analysis and construction of PPI network based on DEGs were accomplished. Combined with detailed clinical information in TCGA database, LASSO and Cox analysis was applied to screen genes closely related to the prognosis of patients in 919 DEGs. In the end, 10 mRNAs were selected to form a prognostic risk assessment model. According to this risk model, patients were allocated into high-risk and low-risk groups in a training set. Survival analysis showed that there was significant difference of OS time between the high-risk and low-risk groups. Subsequently, the risk assessment model was validated in the testing set and the overall cases, and results showed that the model composed of 10 mRNAs could effectively forecast the prognosis of STAD patients. ROC analysis displayed that the AUC value of 5-year survival rate in 3 groups were all greater than 0.75, indicating that the model has great predictive performance. In addition, univariate and multivariate Cox regression analysis of the 3 groups has proven that the model can forecast the prognosis of patients independently. In order to predict the survival rate of patients individually, the risk assessment model and other clinical variables were combined to construct a nomogram. Finally, the pathways in which genes were enriched in high-risk and low-risk groups were identified by GSEA analysis, respectively.
In previous studies, it has been shown that several mRNAs in the risk assessment model can be involved in the development and prognosis of gastric cancer. SLC17A9 has been proven to be highly expressed in gastric cancer, and its high expression is related to a variety of clinical variables, such as advanced TNM stage, T stage, and N stage [26]. In addition, SLC17A9 was found to serve as an independent influencing factor for the survival of gastric cancer patients [26]. It has been found that the high expression of NRP1 was closely related to the malignant phenotype of gastric cancer, and NRP1 could enhance the proliferation, migration, and invasion of gastric cancer cells [27]. NRP1 is also expected to be a therapeutic target for gastric cancer; the study of Zhang et al. showed that the new tumor homing peptide iRGD could enhance the 5-FU chemotherapy effect on gastric cancer through NRP1 [28]. Dong et al. believed that the overexpression of TRIB3 was related to tumor angiogenesis and poor prognosis in gastric cancer patients and TRIB3 was a promising molecular target for antiangiogenic therapy in gastric cancer [29]. Moreover, Wu et al. proposed that the expression of TRIB3 could affect the apoptosis induced by the anticancer drug adriamycin, indicating that TRIB3 expression in response of anticancer drugs may reflect the therapeutic efficacy of gastric cancer [30].
FERMT3 is a member of kindlin family, and several studies have shown that mutation in FERMT3 is an important factor leading to cause leukocyte adhesion deficiency-III (LAD-III) [31–33]. In addition, FERMT3 could promote the proliferation and chemical resistance of glioblastoma cells through integrin mediated Wnt signaling [34]. MAGEA plays a part in various tumors. According to Chen et al., the occurrence and chemoresistance of hepatocellular carcinoma can be regulated by the MAGEA3/LINC01234/miR-31-5p axis [35]. It has been found that MAGEA has a tumor-promoting effect in pancreatic cancer [36], and silence of MAGEA can inhibit the growth of colorectal cancer cells through activating the AMPK pathway [37]. In addition, high expression of MAGEA was demonstrated to be related to poor prognosis of cutaneous squamous cell carcinoma and urothelial bladder cancer [38,39], and MAGEA could work as an independent influencing factor for the prognosis of esophageal squamous cell carcinoma [40]. Nevertheless, the role of the 2 markers in gastric cancer has not been widely explored.
MMRN1 works as a significant part in the blood system. In platelets, MMRN1 acts as a binding protein for coagulation factor V (a key regulator of coagulation), affecting the function and storage of factor V [41]. Besides, Laszlo et al. drew a point that MMRN1 could be used as a new marker to refine the risk stratification of pediatric acute myeloid leukemia [42]. It has been found that RNASE1 has myocardial protective effect [43,44], however, the role of RNASE1 in tumors has not been paid much attention. Previous studies have demonstrated that mutation in PGAP3 is closely related to the occurrence of HPMRS (hyperphosphatasia with mental retardation syndrome) [45–47]. TACR2, also known as NK2R, is a G protein-coupled neurokinin receptor [48]. Tachykinin belongs to the neuropeptide family and is an effective regulator of smooth muscle function. It participates in various physiological and pathological processes of the gastrointestinal tract through 3 different types of receptors (NK1R, NK2R, and NK3R) [48]. Studies have suggested a major involvement of NK2R in the regulation of human colon function [49]. CYP51A1, a member of the cytochrome P450 family, is a key enzyme in cholesterol biosynthesis [50]. Regulating cholesterol biosynthesis is a significant part of the treatment of hypercholesterolemia, and CYP51A1 was demonstrated to be a promising molecular target [51]. The 5 signatures mentioned play roles in different biological processes, but their possible roles in cancer, especially gastric cancer, have hardly been reported. Therefore, the potential role of these genes in cancer and the link between the biological functions they participate in and cancer requires comprehensive research.
Numerous studies have reported many molecular targets that play significant parts in the development and prognosis of gastric cancer, as well as their detailed mechanisms. However, there is still a distance between the results of molecular biological fields and clinical application, and the transformation research is a constant challenge. In recent years, with the rapid development of accurate prediction and individualized treatment, it has become a trend to integrate genomic profiles and clinical characteristics of patients [52]. Similar to this study, previous studies have attempted to combine gene expression profiles with TNM staging to more accurately predict patients’ outcome and improve clinical decision making. For example, G factor, which is a supplement to conventional TNM system, has been demonstrated to be useful in several cancers. G factor based on MMP-7 and p53 has been shown to be a promising index in predicting outcome of stage II/III gastric cancer, and possibly help choose the treatment for patients with stage II [53]. Besides, G factor based on Reg-IV and VEGF-C was considered a promising index for clinical staging of colorectal cancer, and it may be a good indicator of adjuvant chemotherapy for G2 patients in stage II [54]. Therefore, the information of gene expression profiles in gastric cancer microenvironment was associated with patients’ prognosis in this study, which is expected to achieve transformative value in the clinical treatment of gastric cancer.
There were some limitations in this study. First, the study was a retrospective study on the basis of a public database, and the research could not easily cover the differences caused by regional factors. Second, the sample size was not large enough in the current study. In the end, all the samples in this study were obtained from gastric adenocarcinoma, the largest type of gastric cancer. Whether the risk model is applicable to other types of gastric cancer needs further exploration. Thus, the results of this study need a well-designed, multicenter, large sample prospective clinical trial for further verification.
Conclusions
In summary, DEGs were screened based on the gastric cancer microenvironment, and a prognostic risk assessment model composed of 10 mRNAs (SLC17A9, FERMT3, NRP1, MMRN1, RNASE1, TRIB3, PGAP3, MAGEA3, TACR2, and CYP51A1) was constructed. The model can serve as a prognostic index for gastric cancer patients independently, as well as providing supplementary information to improve the prognosis stratification of patients.
Figures
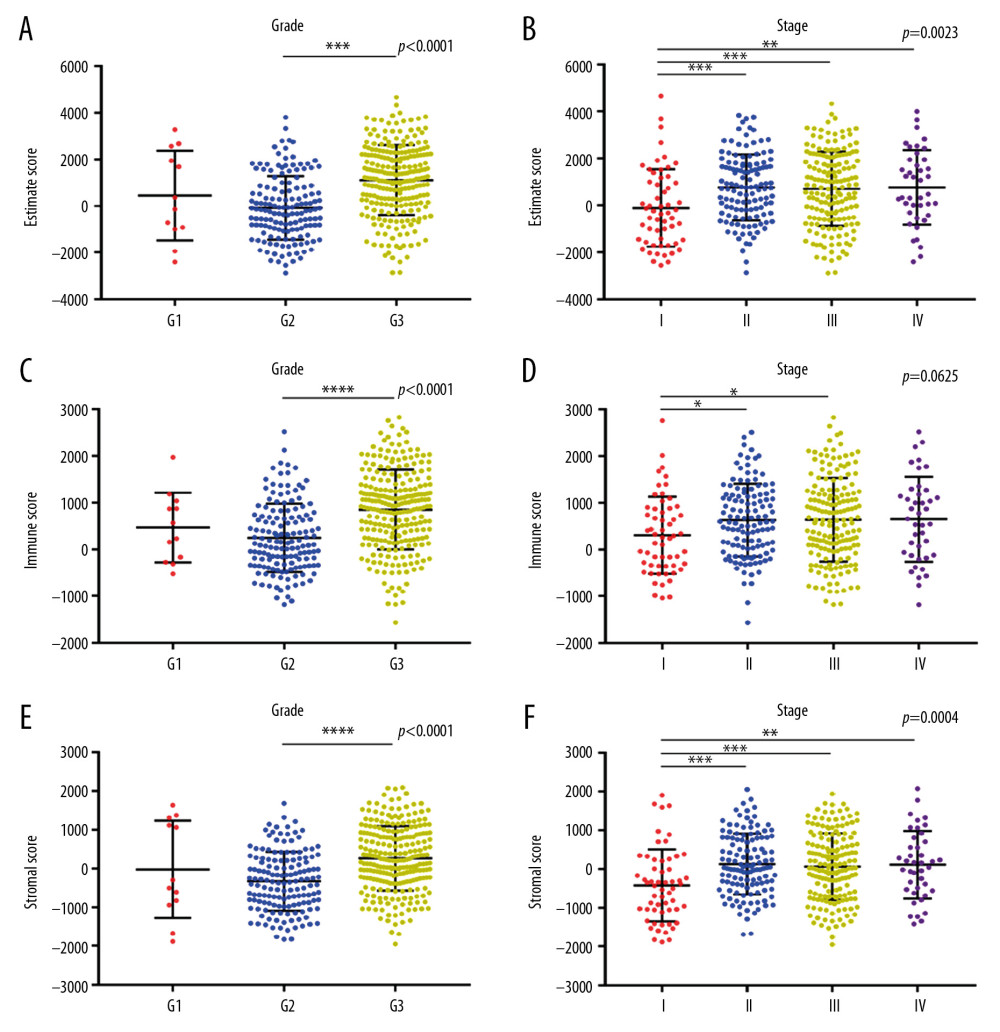 Figure 1. ESTIMATE/immune/stromal scores were associated with grade and stage. (A, B) ESTIMATE score significantly correlated with grade (P<0.0001) and AJCC stage (P=0.0023). (C, D) Immune score was associated with grade significantly (P<0.0001), while showed no correlation with stage (P=0.0625). (E, F) Stromal score had significant relevance with grade (P<0.0001) and stage (P=0.0004).
Figure 1. ESTIMATE/immune/stromal scores were associated with grade and stage. (A, B) ESTIMATE score significantly correlated with grade (P<0.0001) and AJCC stage (P=0.0023). (C, D) Immune score was associated with grade significantly (P<0.0001), while showed no correlation with stage (P=0.0625). (E, F) Stromal score had significant relevance with grade (P<0.0001) and stage (P=0.0004). 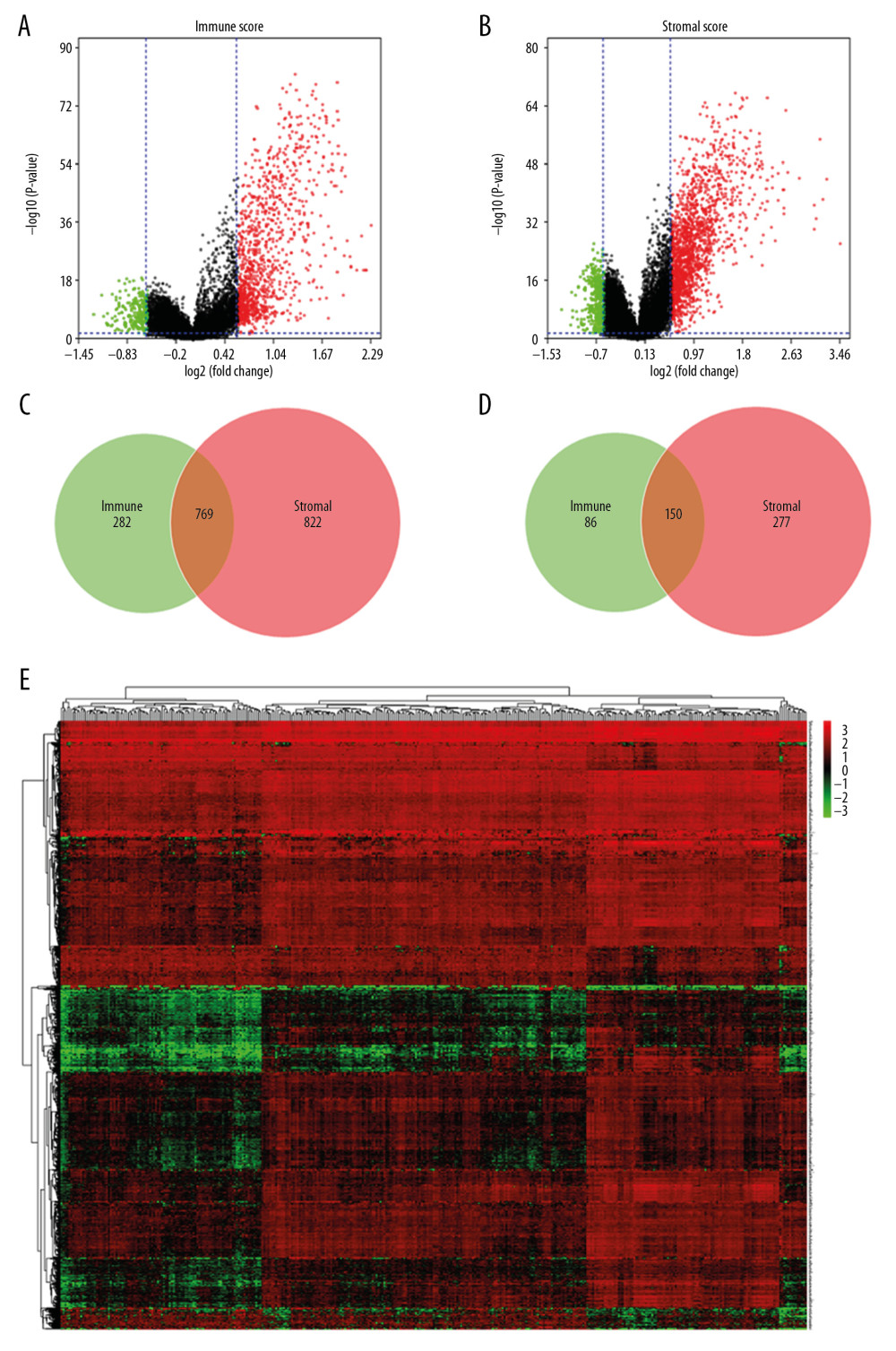 Figure 2. Differentially expressed genes (DEGs) with immune/stromal score in stomach adenocarcinoma (STAD). (A) According to immune score, 1051 genes were high-expressed and 236 genes down-expressed in the high score group compared with the low score group (B) Similarly, 1591 high-expressed genes and 427 down-expressed genes were obtained based on stromal score. (C, D) In total, 769 DEGs were synchronously high-expressed in both the high score groups, while 150 genes were commonly down-expressed. (E) Heat map including 919 DEGs showed distinct gene expression profiles in STAD.
Figure 2. Differentially expressed genes (DEGs) with immune/stromal score in stomach adenocarcinoma (STAD). (A) According to immune score, 1051 genes were high-expressed and 236 genes down-expressed in the high score group compared with the low score group (B) Similarly, 1591 high-expressed genes and 427 down-expressed genes were obtained based on stromal score. (C, D) In total, 769 DEGs were synchronously high-expressed in both the high score groups, while 150 genes were commonly down-expressed. (E) Heat map including 919 DEGs showed distinct gene expression profiles in STAD. 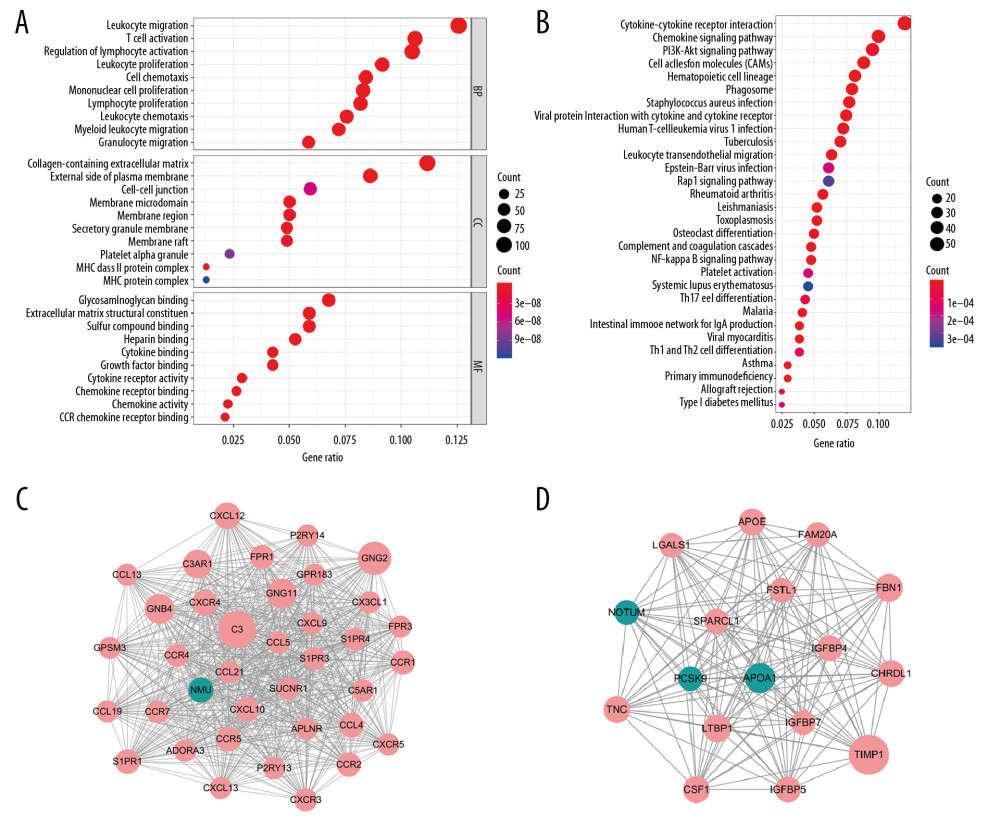 Figure 3. Functional enrichment analysis of 919 DEGs and significant modular analysis based on PPI network. (A) Top 30 GO terms of GO: BP, GO: CC and GO: MF. (B) Top 30 KEGG terms. (C) PPI network of C3 module. (D) PPI network of TIMP1 module. In A, B, terms are sorted by the number of genes enriched. In C, D, red stands for upregulated and green stands for downregulated genes. The size of the node represents the number of proteins that interact with the specified protein. DEGs – different expressed genes; PPI – protein–protein interaction; GO – Gene Ontology; BP – biological processes; CC – cellular components; MF – molecular function; KEGG – Kyoto Encyclopedia of Gene and Genome.
Figure 3. Functional enrichment analysis of 919 DEGs and significant modular analysis based on PPI network. (A) Top 30 GO terms of GO: BP, GO: CC and GO: MF. (B) Top 30 KEGG terms. (C) PPI network of C3 module. (D) PPI network of TIMP1 module. In A, B, terms are sorted by the number of genes enriched. In C, D, red stands for upregulated and green stands for downregulated genes. The size of the node represents the number of proteins that interact with the specified protein. DEGs – different expressed genes; PPI – protein–protein interaction; GO – Gene Ontology; BP – biological processes; CC – cellular components; MF – molecular function; KEGG – Kyoto Encyclopedia of Gene and Genome. 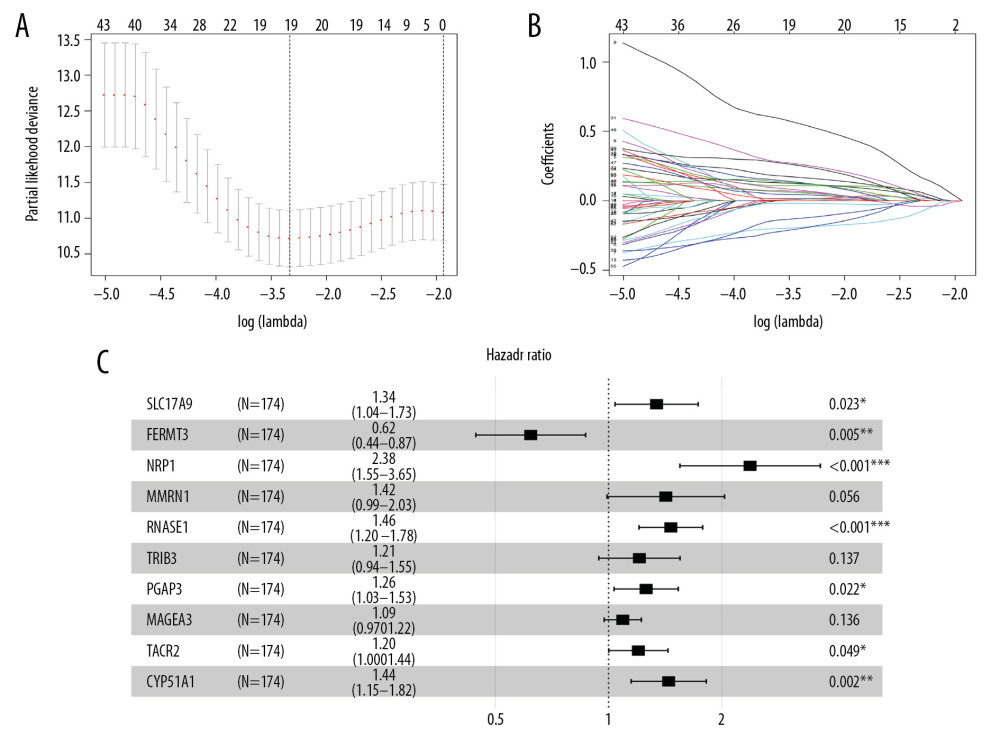 Figure 4. Construction of risk assessment model by LASSO and Cox regression analysis in training group. (A) Selection of the optimal parameter (lambda) in the LASSO model for STAD. (B) LASSO coefficient profiles of genes in STAD. A coefficient profile plot was generated against the log (lambda) sequence. (C) After optimization by Cox analysis, 10 mRNAs were selected to construct the risk assessment model as shown in forest graph. LASSO – least absolute shrinkage and selection operator; STAD – stomach adenocarcinoma.
Figure 4. Construction of risk assessment model by LASSO and Cox regression analysis in training group. (A) Selection of the optimal parameter (lambda) in the LASSO model for STAD. (B) LASSO coefficient profiles of genes in STAD. A coefficient profile plot was generated against the log (lambda) sequence. (C) After optimization by Cox analysis, 10 mRNAs were selected to construct the risk assessment model as shown in forest graph. LASSO – least absolute shrinkage and selection operator; STAD – stomach adenocarcinoma. 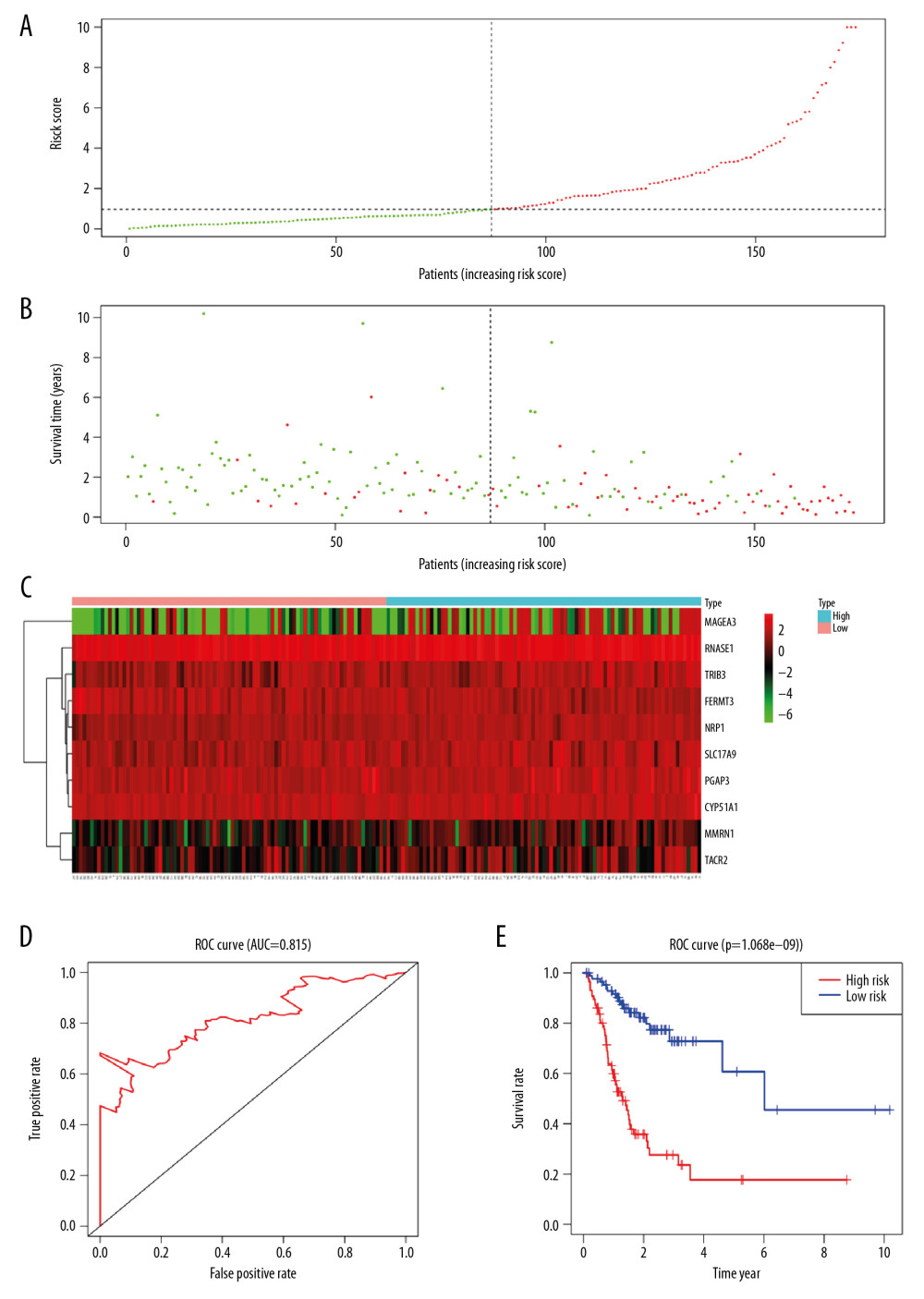 Figure 5. Characteristics of the risk assessment model in training group. (A, B) The distribution of risk score, patients’ survival and status for STAD. The black dotted line divided patients into high-risk group and low-risk group. (C) Heatmap of gene expression profiles in risk assessment model for STAD. (D) The ROC curve was applied to assess the predictive performance of the risk assessment model. (E) Kaplan-Meier survival analysis of STAD patients stratified by the median risk score. STAD – stomach adenocarcinoma; ROC – receiver operating characteristic.
Figure 5. Characteristics of the risk assessment model in training group. (A, B) The distribution of risk score, patients’ survival and status for STAD. The black dotted line divided patients into high-risk group and low-risk group. (C) Heatmap of gene expression profiles in risk assessment model for STAD. (D) The ROC curve was applied to assess the predictive performance of the risk assessment model. (E) Kaplan-Meier survival analysis of STAD patients stratified by the median risk score. STAD – stomach adenocarcinoma; ROC – receiver operating characteristic. 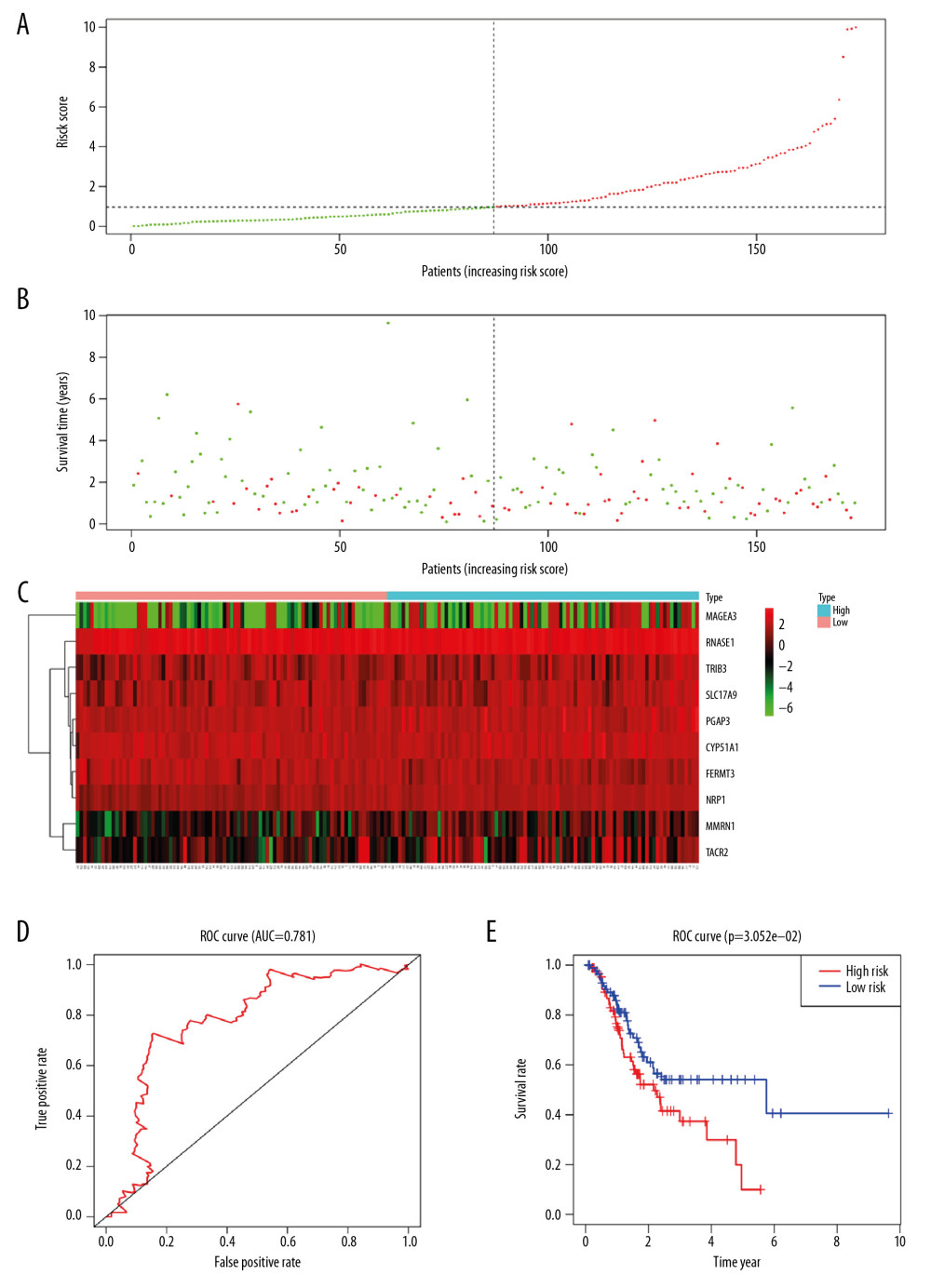 Figure 6. Validation of risk assessment model in testing set. The distribution of risk score (A), survival time (B), heatmap (C), ROC curve (D) and Kaplan Meier survival curve (E) for the testing set. ROC – receiver operating characteristic.
Figure 6. Validation of risk assessment model in testing set. The distribution of risk score (A), survival time (B), heatmap (C), ROC curve (D) and Kaplan Meier survival curve (E) for the testing set. ROC – receiver operating characteristic. 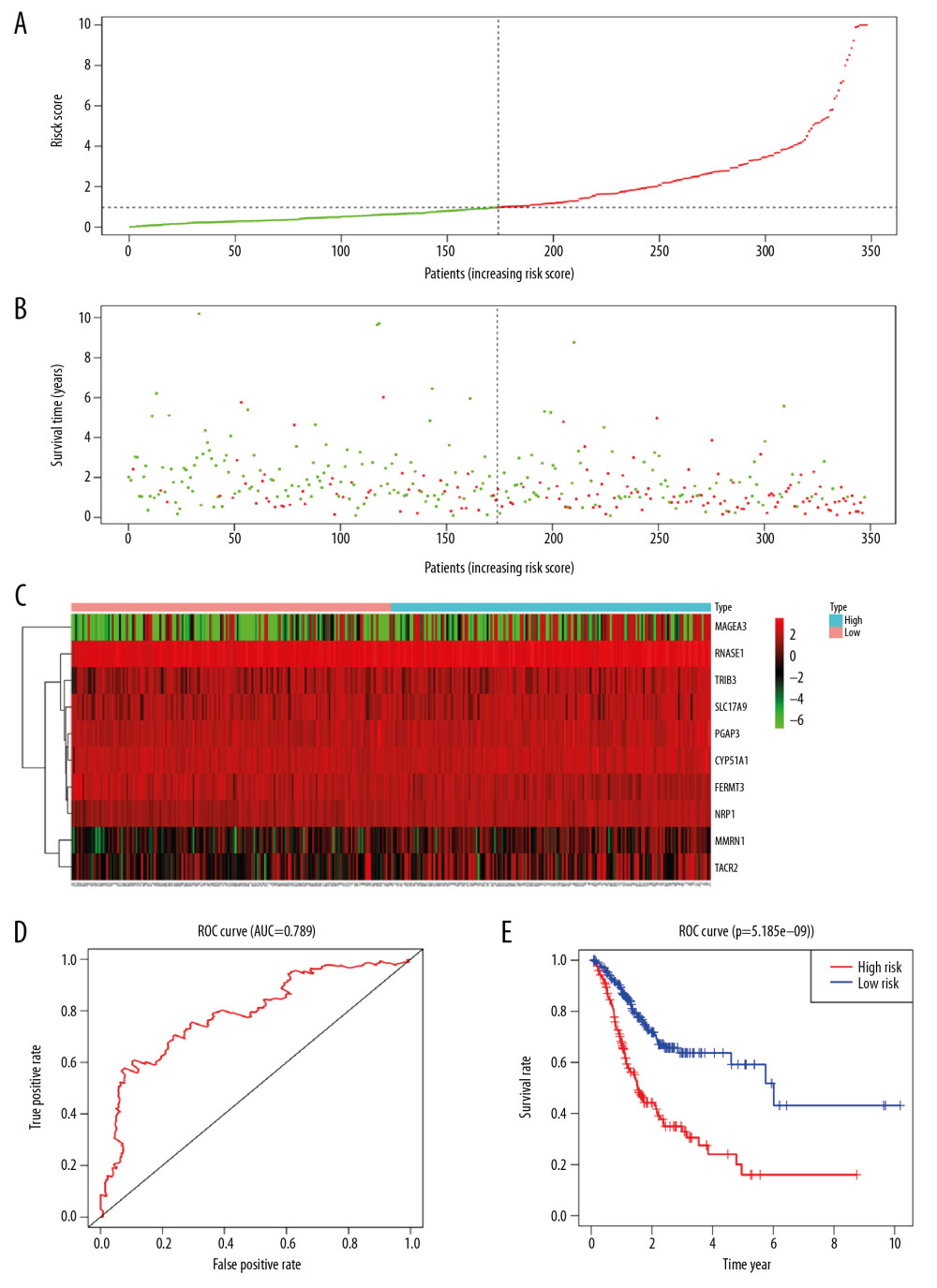 Figure 7. Validation of risk assessment model in overall patients cohort. The distribution of risk score (A), survival time (B), heatmap (C), ROC curve (D), and Kaplan Meier survival curve (E) for the overall patients. ROC – receiver operating characteristic.
Figure 7. Validation of risk assessment model in overall patients cohort. The distribution of risk score (A), survival time (B), heatmap (C), ROC curve (D), and Kaplan Meier survival curve (E) for the overall patients. ROC – receiver operating characteristic. 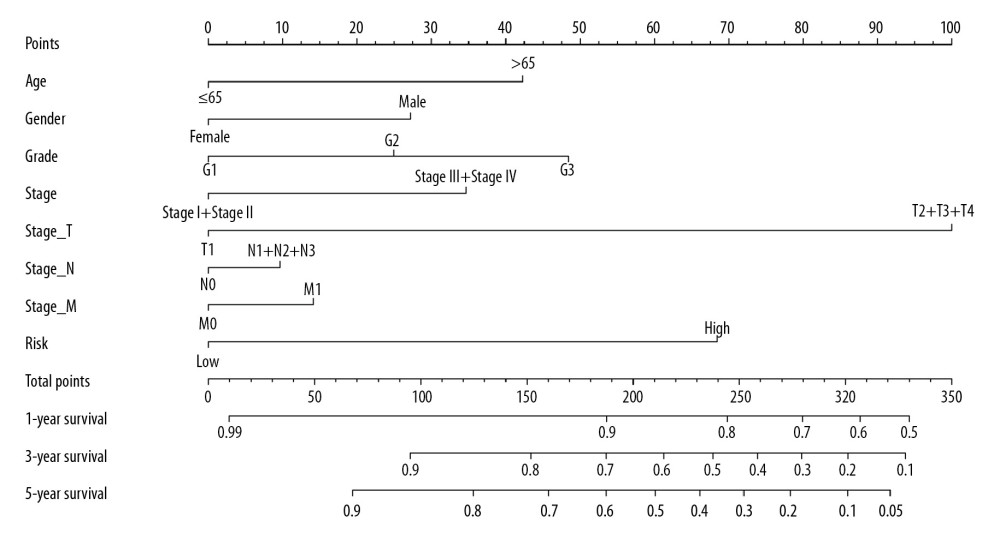 Figure 8. The nomogram to predict the probability of 1-year, 3-year, and 5-year survival rate in stomach adenocarcinoma.
Figure 8. The nomogram to predict the probability of 1-year, 3-year, and 5-year survival rate in stomach adenocarcinoma. 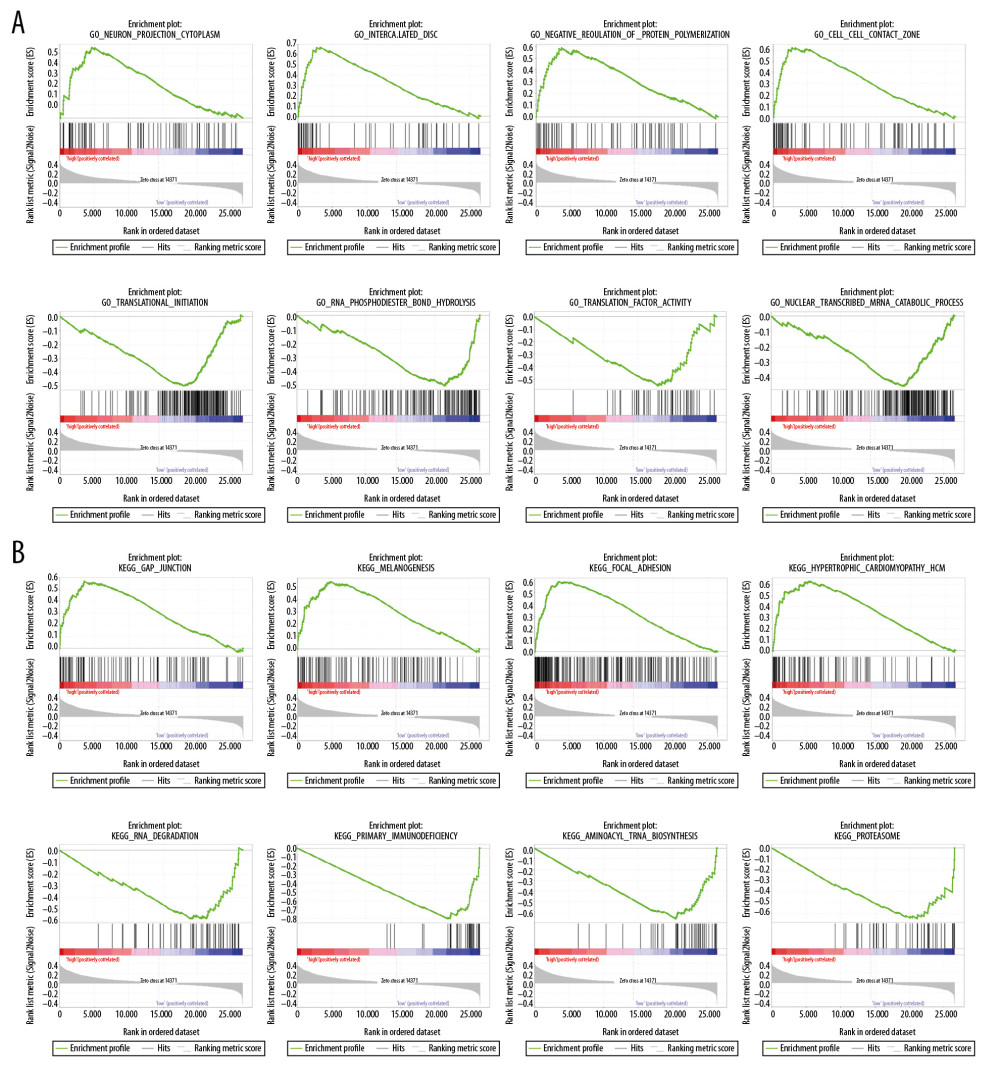 Figure 9. GSEA analysis in GO gene set (A) and KEGG gene set (B). GSEA – Gene Set Enrichment Analysis; GO – Gene Ontology; KEGG – Kyoto Encyclopedia of Gene and Genome.
Figure 9. GSEA analysis in GO gene set (A) and KEGG gene set (B). GSEA – Gene Set Enrichment Analysis; GO – Gene Ontology; KEGG – Kyoto Encyclopedia of Gene and Genome. References
1. Sun Z, Wang ZN, Zhu Z, Evaluation of the seventh edition of American Joint Committee on Cancer TNM staging system for gastric cancer: Results from a Chinese mono-institutional study: Ann Surg Oncol, 2012; 19; 1918-27
2. Zheng L, Wu C, Xi P, The survival and the long-term trends of patients with gastric cancer in Shanghai, China: BMC Cancer, 2014; 14; 300
3. Wang W, Zheng C, Fang C, Time trends of clinicopathologic features and surgical treatment for gastric cancer: Results from 2 high-volume institutions in southern China: Surgery, 2015; 158; 1590-97
4. Sano T, Coit DG, Kim HH, Proposal of a new stage grouping of gastric cancer for TNM classification: International Gastric Cancer Association staging project: Gastric Cancer, 2017; 20; 217-25
5. Edge SB, Compton CC: Ann Surg Oncol, 2010; 17; 1471-74
6. Amin MB, Greene FL, Edge SB, The Eighth Edition AJCC Cancer Staging Manual: Continuing to build a bridge from a population-based to a more “personalized” approach to cancer staging: Cancer J Clin, 2017; 67; 93-99
7. Fang WL, Huang KH, Chen JH: World J Surg, 2011; 35; 2723-29
8. Fang C, Wang W, Deng JY: Cancer Commun (Lond), 2018; 38; 67
9. Wang H, Guo W, Hu Y: Mol Clin Oncol, 2018; 9; 423-31
10. Graziosi L, Marino E, Donini A: Eur J Surg Oncol, 2019; 45; 1105-8
11. Chae S, Lee A, Lee JH: Gastric Cancer, 2011; 14; 166-71
12. Bang YJ, Kim YW, Yang HK, Adjuvant capecitabine and oxaliplatin for gastric cancer after D2 gastrectomy (CLASSIC): A phase 3 open-label, randomised controlled trial: Lancet, 2012; 379; 315-21
13. Noh SH, Park SR, Yang HK, Adjuvant capecitabine plus oxaliplatin for gastric cancer after D2 gastrectomy (CLASSIC): 5-year follow-up of an open-label, randomised phase 3 trial: Lancet Oncol, 2014; 15; 1389-96
14. Hanahan D, Coussens LM, Accessories to the crime: functions of cells recruited to the tumor microenvironment: Cancer Cell, 2012; 21; 309-22
15. Hanahan D, Weinberg RA, Hallmarks of cancer: The next generation: Cell, 2011; 144; 646-74
16. Jia D, Li S, Li D, Mining TCGA database for genes of prognostic value in glioblastoma microenvironment: Aging (Albany NY), 2018; 10; 592-605
17. Xu WH, Xu Y, Wang J, Prognostic value and immune infiltration of novel signatures in clear cell renal cell carcinoma microenvironment: Aging (Albany NY), 2019; 11; 6999-7020
18. Pietras K, Östman A, Hallmarks of cancer: Interactions with the tumor stroma: Exp Cell Res, 2010; 316; 1324-31
19. Chung HW, Lim JB, Role of the tumor microenvironment in the pathogenesis of gastric carcinoma: World J Gastroenterol, 2014; 20; 1667-80
20. Wang J, Li D, Cang H, Guo B, Crosstalk between cancer and immune cells: Role of tumor-associated macrophages in the tumor microenvironment: Cancer Med, 2019; 8; 4709-21
21. Guo S, Deng CX, Effect of stromal cells in tumor microenvironment on metastasis initiation: Int J Biol Sci, 2018; 14; 2083-93
22. Garcia-Gomez A, Rodriguez-Ubreva J, Ballestar E, Epigenetic interplay between immune, stromal and cancer cells in the tumor microenvironment: Clin Immunol, 2018; 196; 64-71
23. Cheng HS, Lee J, Wahli W, Tan NS, Exploiting vulnerabilities of cancer by targeting nuclear receptors of stromal cells in tumor microenvironment: Mol Cancer, 2019; 18; 51
24. Marzagalli M, Ebelt ND, Manuel ER, Unraveling the crosstalk between melanoma and immune cells in the tumor microenvironment: Semin Cancer Biol, 2019; 59; 236-50
25. Yoshihara K, Shahmoradgoli M, Martinez E, Inferring tumour purity and stromal and immune cell admixture from expression data: Nat Commun, 2013; 4; 2612
26. Li J, Su T, Yang L, High SLC17A9 expression correlates with poor survival in gastric carcinoma: Future Oncol, 2019; 15(36); 4155-66
27. Wang G, Shi B, Fu Y, Hypomethylated gene NRP1 is co-expressed with PDGFRB and associated with poor overall survival in gastric cancer patients: Biomed Pharmacother, 2019; 111; 1334-41
28. Zhang L, Xing Y, Gao Q, Combination of NRP1-mediated iRGD with 5-fluorouracil suppresses proliferation, migration and invasion of gastric cancer cells: Biomed Pharmacother, 2017; 93; 1136-43
29. Dong S, Xia J, Wang H, Overexpression of TRIB3 promotes angiogenesis in human gastric cancer: Oncol Rep, 2016; 36; 2339-48
30. Wu IJ, Lin RJ, Wang HC, TRIB3 downregulation enhances doxorubicin-induced cytotoxicity in gastric cancer cells: Arch Biochem Biophys, 2017; 622; 26-35
31. Shahid S, Zaidi S, Ahmed S, A novel nonsense mutation in FERMT3 causes LAD-III in a Pakistani family: Front Genet, 2019; 10; 360
32. Suratannon N, Yeetong P, Srichomthong C, Adaptive immune defects in a patient with leukocyte adhesion deficiency type III with a novel mutation in FERMT3: Pediatr Allergy Immunol, 2016; 27; 214-17
33. Jurk K, Schulz AS, Kehrel BE, Novel integrin-dependent platelet malfunction in siblings with leukocyte adhesion deficiency-III (LAD-III) caused by a point mutation in FERMT3: Thromb Haemost, 2010; 103; 1053-64
34. Lu C, Cui C, Liu B, FERMT3 contributes to glioblastoma cell proliferation and chemoresistance to temozolomide through integrin mediated Wnt signaling: Neurosci Lett, 2017; 657; 77-83
35. Chen Y, Zhao H, Li H, LINC01234/MicroRNA-31-5p/MAGEA3 axis mediates the proliferation and chemoresistance of hepatocellular carcinoma cells: Mol Ther Nucleic Acids, 2019; 19; 168-78
36. Das B, Senapati S, Functional and mechanistic studies reveal MAGEA3 as a pro-survival factor in pancreatic cancer cells: J Exp Clin Cancer Res, 2019; 38; 294
37. Wu F, Liu F, Dong L, miR-1273g silences MAGEA3/6 to inhibit human colorectal cancer cell growth via activation of AMPK signaling: Cancer Lett, 2018; 435; 1-9
38. Abikhair M, Roudiani N, Mitsui H, MAGEA3 expression in cutaneous squamous cell carcinoma is associated with advanced tumor stage and poor prognosis: J Invest Dermatol, 2017; 137; 775-78
39. Lausenmeyer EM, Braun K, Breyer J, Strong expression of cancer test is antigens CTAG1B and MAGEA3 is correlated with unfavourable histopathological features and MAGEA3 is associated with worse progression-free survival in urothelial bladder cancer: Urol Int, 2019; 102; 77-82
40. Liu S, Chen H, Ge X, MAGEA3 serves as an independent indicator for predicting the prognosis of ESCC: Panminerva Med, 2019 [Online ahead of print]
41. Jeimy SB, Krakow EF, Fuller N, An acquired factor V inhibitor associated with defective factor V function, storage and binding to multimerin 1: J Thromb Haemost, 2008; 6; 395-97
42. Laszlo GS, Alonzo TA, Gudgeon CJ, Multimerin-1 (MMRN1) as novel adverse marker in pediatric acute myeloid leukemia: A report from the Children’s Oncology Group: Clin Cancer Res, 2015; 21; 3187-95
43. Cabrera-Fuentes HA, Niemann B, Grieshaber P, RNase1 as a potential mediator of remote ischaemic preconditioning for cardioprotection dagger: Eur J Cardiothorac Surg, 2015; 48; 732-37
44. Cabrera-Fuentes HA, Ruiz-Meana M, Simsekyilmaz S, RNase1 prevents the damaging interplay between extracellular RNA and tumour necrosis factor-alpha in cardiac ischaemia/reperfusion injury: Thromb Haemost, 2014; 112; 1110-19
45. Howard MF, Murakami Y, Pagnamenta AT, Mutations in PGAP3 impair GPI-anchor maturation, causing a subtype of hyperphosphatasia with mental retardation: Am J Hum Genet, 2014; 94; 278-87
46. Abdel-Hamid MS, Issa MY, Otaify GA, PGAP3-related hyperphosphatasia with mental retardation syndrome: Report of 10 new patients and a homozygous founder mutation: Clin Genet, 2018; 93; 84-91
47. Knaus A, Awaya T, Helbig I, Rare noncoding mutations extend the mutational spectrum in the PGAP3 subtype of hyperphosphatasia with mental retardation syndrome: Hum Mutat, 2016; 37; 737-44
48. Zhang K, Chen QT, Li JH, The expression of tachykinin receptors in the human lower esophageal sphincter: Eur J Pharmacol, 2016; 774; 144-49
49. Jaafari N, Hua G, Adelaide J, Expression of the tachykinin receptor mRNAs in healthy human colon: Eur J Pharmacol, 2008; 599; 121-25
50. Pickl JM, Kamel W, Ciftci S, Opposite expression of CYP51A1 and its natural antisense transcript AluCYP51A1 in adenovirus type 37 infected retinal pigmented epithelial cells: Febs Lett, 2015; 589; 1383-88
51. Kaluzhsiy LA, Gnedenko OV, Gilep AAThe screening of the inhibitors of the human cytochrome P450(51) (CYP51A1): The plant and animal structural lanosterol’s analogs: Biomed Khim, 2014; 60; 528-37 [in Russian]
52. Wang H, Wu X, Chen Y, Stromal-immune score-based gene signature: A prognosis stratification tool in gastric cancer: Front Oncol, 2019; 9; 1212
53. Sawada T, Yashiro M, Sentani K, New molecular staging with G-factor supplements TNM classification in gastric cancer: A multicenter collaborative research by the Japan Society for Gastroenterological Carcinogenesis G-Project committee: Gastric Cancer, 2015; 18; 119-28
54. Sawada T, Yashiro M, Sentani K, New molecular staging with G-factors (VEGF-C and Reg IV) by supplementing TNM classification in colorectal cancers: Oncol Rep, 2013; 30; 2609-16
Figures
Figure 1. ESTIMATE/immune/stromal scores were associated with grade and stage. (A, B) ESTIMATE score significantly correlated with grade (P<0.0001) and AJCC stage (P=0.0023). (C, D) Immune score was associated with grade significantly (P<0.0001), while showed no correlation with stage (P=0.0625). (E, F) Stromal score had significant relevance with grade (P<0.0001) and stage (P=0.0004).Figure 2. Differentially expressed genes (DEGs) with immune/stromal score in stomach adenocarcinoma (STAD). (A) According to immune score, 1051 genes were high-expressed and 236 genes down-expressed in the high score group compared with the low score group (B) Similarly, 1591 high-expressed genes and 427 down-expressed genes were obtained based on stromal score. (C, D) In total, 769 DEGs were synchronously high-expressed in both the high score groups, while 150 genes were commonly down-expressed. (E) Heat map including 919 DEGs showed distinct gene expression profiles in STAD.Figure 3. Functional enrichment analysis of 919 DEGs and significant modular analysis based on PPI network. (A) Top 30 GO terms of GO: BP, GO: CC and GO: MF. (B) Top 30 KEGG terms. (C) PPI network of C3 module. (D) PPI network of TIMP1 module. In A, B, terms are sorted by the number of genes enriched. In C, D, red stands for upregulated and green stands for downregulated genes. The size of the node represents the number of proteins that interact with the specified protein. DEGs – different expressed genes; PPI – protein–protein interaction; GO – Gene Ontology; BP – biological processes; CC – cellular components; MF – molecular function; KEGG – Kyoto Encyclopedia of Gene and Genome.Figure 4. Construction of risk assessment model by LASSO and Cox regression analysis in training group. (A) Selection of the optimal parameter (lambda) in the LASSO model for STAD. (B) LASSO coefficient profiles of genes in STAD. A coefficient profile plot was generated against the log (lambda) sequence. (C) After optimization by Cox analysis, 10 mRNAs were selected to construct the risk assessment model as shown in forest graph. LASSO – least absolute shrinkage and selection operator; STAD – stomach adenocarcinoma.Figure 5. Characteristics of the risk assessment model in training group. (A, B) The distribution of risk score, patients’ survival and status for STAD. The black dotted line divided patients into high-risk group and low-risk group. (C) Heatmap of gene expression profiles in risk assessment model for STAD. (D) The ROC curve was applied to assess the predictive performance of the risk assessment model. (E) Kaplan-Meier survival analysis of STAD patients stratified by the median risk score. STAD – stomach adenocarcinoma; ROC – receiver operating characteristic.Figure 6. Validation of risk assessment model in testing set. The distribution of risk score (A), survival time (B), heatmap (C), ROC curve (D) and Kaplan Meier survival curve (E) for the testing set. ROC – receiver operating characteristic.Figure 7. Validation of risk assessment model in overall patients cohort. The distribution of risk score (A), survival time (B), heatmap (C), ROC curve (D), and Kaplan Meier survival curve (E) for the overall patients. ROC – receiver operating characteristic.Figure 8. The nomogram to predict the probability of 1-year, 3-year, and 5-year survival rate in stomach adenocarcinoma.Figure 9. GSEA analysis in GO gene set (A) and KEGG gene set (B). GSEA – Gene Set Enrichment Analysis; GO – Gene Ontology; KEGG – Kyoto Encyclopedia of Gene and Genome. Tables
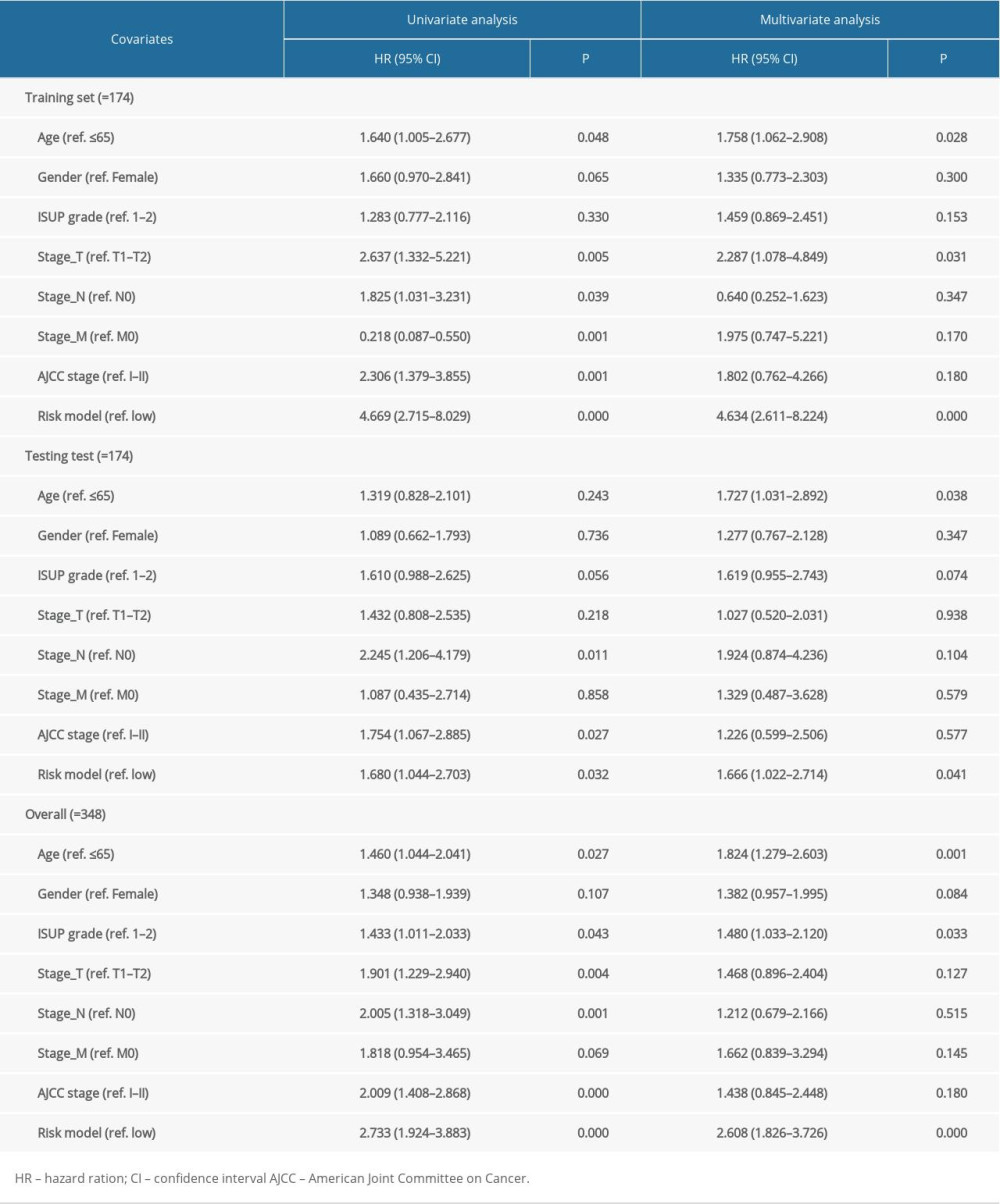 Table 1. Univariate and multivariate Cox regression analysis of the training set, testing set, and overall patients group.Table 1. Univariate and multivariate Cox regression analysis of the training set, testing set, and overall patients group.
Table 1. Univariate and multivariate Cox regression analysis of the training set, testing set, and overall patients group.Table 1. Univariate and multivariate Cox regression analysis of the training set, testing set, and overall patients group.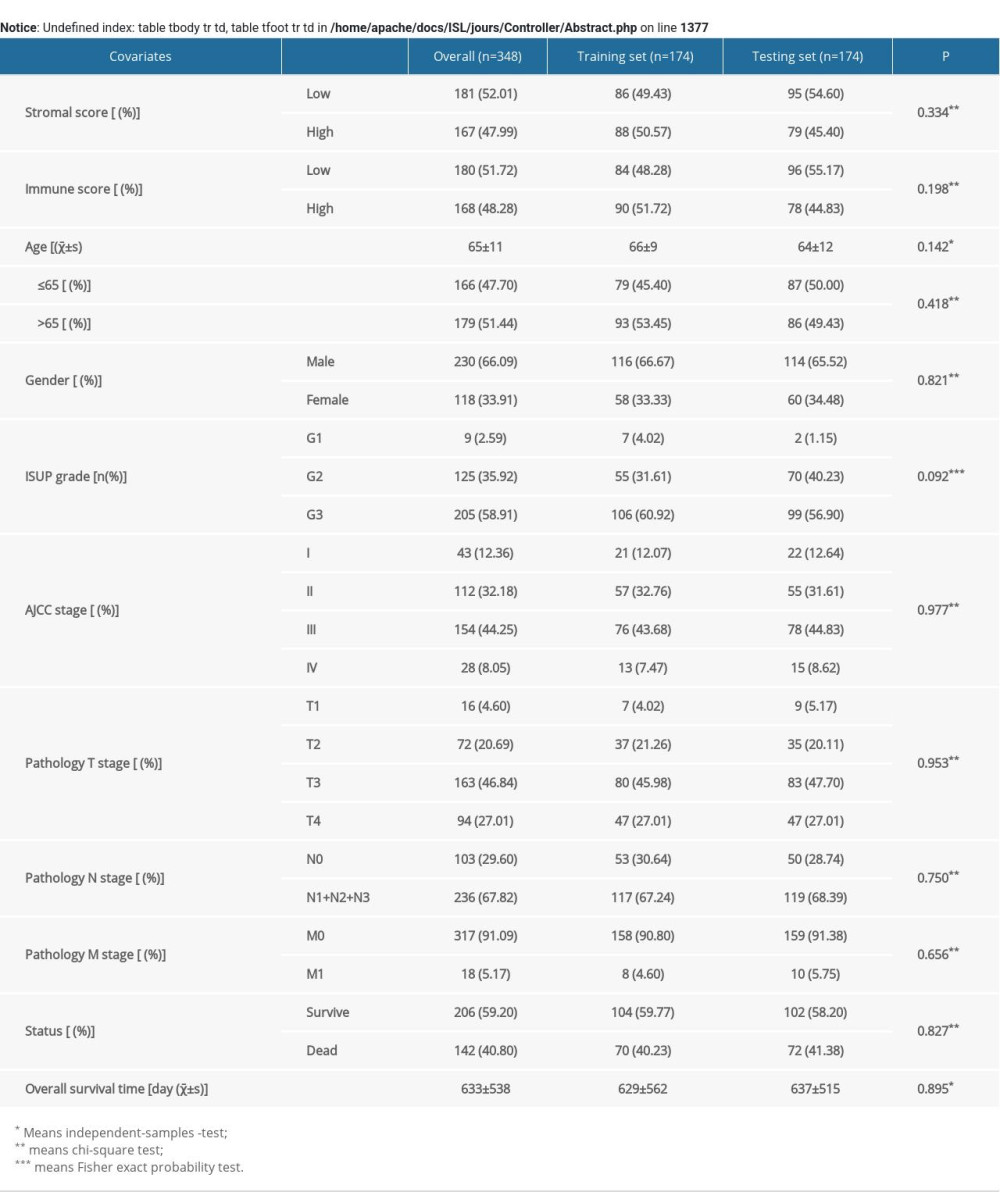 Supplementary Table 1. Basic clinicopathological data of STAD patients.
Supplementary Table 1. Basic clinicopathological data of STAD patients. In Press
Clinical Research
Effects of Single-Bout Endurance Exercise Intensity on Peripheral Neurotrophic Factors in Patients With Isc...Med Sci Monit In Press; DOI: 10.12659/MSM.952089
Review article
Anisodus tanguticus in Cancer Research: A Review of Traditional Use, Phytochemistry, Extraction Methods, an...Med Sci Monit In Press; DOI: 10.12659/MSM.952999
Clinical Research
Nasal Mucociliary Clearance and Its Relationship With Disease Severity in Patients With Multiple SclerosisMed Sci Monit In Press; DOI: 10.12659/MSM.952850
Clinical Research
Modified Thoracoabdominal Nerves Block Through the Perichondrial Approach vs Subcostal Transversus Abdomini...Med Sci Monit In Press; DOI: 10.12659/MSM.953976
Most Viewed Current Articles
17 Jan 2024 : Review article 14,176,570
Vaccination Guidelines for Pregnant Women: Addressing COVID-19 and the Omicron VariantDOI :10.12659/MSM.942799
Med Sci Monit 2024; 30:e942799
13 Nov 2021 : Clinical Research 3,762,188
Acceptance of COVID-19 Vaccination and Its Associated Factors Among Cancer Patients Attending the Oncology ...DOI :10.12659/MSM.932788
Med Sci Monit 2021; 27:e932788
14 Dec 2022 : Clinical Research 2,466,310
Prevalence and Variability of Allergen-Specific Immunoglobulin E in Patients with Elevated Tryptase LevelsDOI :10.12659/MSM.937990
Med Sci Monit 2022; 28:e937990
16 May 2023 : Clinical Research 708,927
Electrophysiological Testing for an Auditory Processing Disorder and Reading Performance in 54 School Stude...DOI :10.12659/MSM.940387
Med Sci Monit 2023; 29:e940387