30 August 2020: Animal Study
Promoting Role of Long Non-Coding RNA Small Nucleolar RNA Host Gene 15 (SNHG15) in Neuronal Injury Following Ischemic Stroke via the MicroRNA-18a/CXC Chemokine Ligand 13 (CXCL13)/ERK/MEK Axis
Tiezhu Guo1AB, Yueting Liu2CE, Xinliang Ren1DE, Wei Wang1EF, Hanrui Liu3F*DOI: 10.12659/MSM.923610
Med Sci Monit 2020; 26:e923610






Abstract
BACKGROUND: Long-non-coding RNA (lncRNA) SNHG15 has been reported to be an aberrantly expressed lncRNA in patients with ischemic stroke, but its role in neuronal injury following ischemic stroke remains unclear. We hypothesized that this lncRNA is associated with the pathogenesis of ischemic stroke.
MATERIAL AND METHODS: A mouse model of ischemic stroke was established by middle cerebral artery occlusion (MCAO). A neurogenic mouse cell line Neuro-2a (N2a) was subjected to oxygen-glucose deprivation (OGD) for in vitro experiments. Expression of SNHG15, microRNA-18a (miR-18a), and CXCL13 in mouse brain and in OGD-treated N2a cells was determined. Altered expression of SNHG15 and miR-18a was introduced to detect their roles in N2a cell viability and apoptosis. Targeting relationships between miR-18a and SNHG15 or CXCL13 were validated by luciferase assays. Cells were treated with the ERK/MEK antagonist U0126 to assess the role of the ERK/MEK signaling pathway in N2a cell growth.
RESULTS: SNHG15 and CXCL13 were overexpressed and miR-18a was underexpressed in MCAO-induced mice and OGD-treated N2a cells. Silencing of SNHG15 or overexpression of miR-18a promoted cell viability, while decreased cell apoptosis induced by OGD; however, subsequent disruption of the ERK/MEK signaling pathway reversed these effects. SNHG15 was found to bind to miR-18a, which could further target CXCL13.
CONCLUSIONS: Silencing of SNHG15 led to CXCL13 upregulation through sequestering miR-18a and the following ERK/MEK activation, thus enhancing viability while reducing apoptosis of N2a cells. SNHG15 may serve as a novel target for ischemic stroke treatment.
Keywords: Chemokine CXCL13, Stroke, Brain Ischemia, Butadienes, Mitogen-Activated Protein Kinases, Nitriles, Protein Kinase Inhibitors, RNA, Small Nucleolar
Background
The brain is the organ with the highest sensitivity to hypoxia [1]. Ischemic stroke, which is an episode of neurological impairment caused by cerebral infarction in the perfusion territory of an occluded or stenosed artery, is a major cause of morbidity and mortality worldwide [2]. The subsequent nutrient insufficiency triggers a series of pathophysiological events, including energy supply failure, inflammation, oxidative stress, and apoptosis, eventually resulting in neuronal cell death [3]. Currently, thrombolytic therapy with tissue plasminogen activator, which can rescue cells in the ischemic penumbra, is the major globally approved treatment for ischemia; however, several problems are still unaddressed, such as the limited therapeutic time window and risk of cerebral hemorrhage [4]. In addition, the reperfusion process during ischemic stroke treatment can lead to serious brain dysfunction [1]. Persistent cell death has been well-established as a significant attribute of ischemic stroke [5]. Thus, identifying novel therapeutic strategies to reduce cell death and to alleviate ischemic stroke is of great importance.
Gene therapy has emerged as a potential therapeutic option for ischemic stroke. RNA composes at least 75% of human genomic sequences, and both protein-coding RNAs (mainly mRNAs) and non-coding RNAs (ncRNAs) exert key functions in multiple cell types and have been developed in current computational and experimental approaches [6]. Long non-coding RNAs (lncRNAs) and microRNAs (miRNAs) are 2 main classes of ncRNAs that regulate fundamental cellular processes through diverse mechanisms [7]. The involvement of aberrant miRNA and lncRNA expression in ischemic stroke has attracted much attention [8]. miRNAs are well known to produce gene degradation by binding to the 3’untranslated region (3’UTR) of target mRNAs, but these repressive effects can be diminished by other transcripts, including lncRNAs, through the shared response elements, which are termed competitive endogenous RNA (ceRNA) networks [9]. Several such networks have been validated to fulfill key functions in neuron apoptosis and nerve injury after ischemic stroke [10,11]. Small nucleolar RNA host gene 15 (SNHG15) is a strongly conserved lncRNA located at 7p13 with 860 bp in length, and its functions in tumor progression in several human cancers have been well documented [12]. SNHG15 has been noted to promote glioma progression [13]. Interestingly, a high expression profile of SNHG15 has been found in patients with ischemic stroke, indicating the promising treatment and prognostic values of this lncRNA [14]. However, the exact role of SNHG15 in ischemic stroke or other neurological diseases, to the best of our knowledge, has not been previously explored. Intriguingly, our study confirmed SNHG15 as a sponge for miR-18a. miR-18a is a member of the miR-17-92 cluster (containing miR-17, miR18a, miR-19a, miR-19b miR-20a, and miR-92a), whose dysregulation was mainly found to be linked to cancer and to other human diseases such as ischemic stroke [15]. miR-18a has been suggested to be a promising therapeutic agent to improve the function of cerebral arteriovenous malformation endothelial cells to relieve neurological impairment [16], but its role in ischemic stroke is unclear. In addition, CXC chemokine ligand 13 (CXCL13), which has been reported to play a key role in central nervous system (CNS) inflammation under a wide array of circumstances [17], was identified as a target mRNA of miR-18a. Therefore, we hypothesized that SNHG15 affects neurological functions after ischemic stroke through the miR-18a/CXCL13 network. Through both animal and cell experiments, we demonstrated that silencing of SNHG15 improved the neurological function and decreased the neuron loss following ischemic stroke through upregulation of miR-18a and downregulation of CXCL13. Activation of the ERK/MEK signaling pathway was also found to be involved in these events.
Material and Methods
ETHICS STATEMENT:
This study was ratified by the Clinical Ethics Committee of Heji Hospital Affiliated with Changzhi Medical College. All experimental procedures were conducted in accordance with the ethics guidelines for the study of experimental pain in conscious animals. Great attempts were made to minimize the number and suffering of animals.
ESTABLISHMENT OF A MOUSE MODEL WITH ISCHEMIC STROKE:
A total of 16 male C57BL.6J mice (8–10 weeks old, 23–27 g) were purchased from SLAC Laboratory Animal Co. (Shanghai, China). A mouse model of ischemic stroke was induced by middle cerebral artery occlusion (MCAO) as previously described [18]. In brief, after 1 week of acclimatization with free access to food and water and a 12-h dark/light cycle, the mice were numbered by weight and randomly allocated into a sham group (n=8) and an MCAO model group (n=8). The mice were anaesthetized with a mixture of oxygen and isoflurane (5% for induction and 1.5% for maintenance). Then, the mice were depilated and placed on a heating pad in an upright position. The near end of the right common carotid artery (RccA) and carotid artery was ligated using a 6-0 polyester suture. MCAO was introduced at the far end, 10 mm from the bifurcation of internal carotid artery. Then, the far end of the internal carotid artery node was ligated and maintained. After 30 min of ischemia, the ligation was removed, followed by reperfusion. After recovering from anesthesia, the mice were given water and food. Mice in the sham group only had the median neck opened, and the RccA was cut open and then sutured. Twenty-four hours after reperfusion, the neurologic deficit of mice was scored using the criteria by Longa et al. [19]. Then, the animals were instantly euthanized and the brain tissues were collected for subsequent experiments. The brain tissue sections from 4 mice were used for TTC staining. The left hemispheres of the remining 4 mice were used for protein and RNA examinations, while the right hemispheres were used for histological staining.
After neurologic deficit scoring, the animals were instantly euthanized through injection of an overdose of pentobarbital (100 mg/kg), and animal death was confirmed by respiratory and cardiac arrest and loss of cerebellum and nerve reflex. Then, the mouse brains were taken out and the hippocampal tissues were cut into 5 coronal sections (2-mm). The tissues were stained with 2,3,5-triphenyltetrazolium chloride (TTC, Sigma-Aldrich Chemical Company, WI, USA). The lesion area was measured using Image J software (v1.52r, National Institutes of Health, Bethesda, MD, USA) to evaluate the infarct size. Moreover, the caspase-3 expression in the hippocampus was detected using immunohistochemistry. In brief, the hippocampal tissue sections were dewaxed, hydrated with an ethanol series, warmed in citrate buffer for antigen retrieval, washed, blocked, and incubated with normal goat serum (#E510009, Sangon Biotech Co., Shanghai, China), and then incubated with rabbit anti-mouse caspase-3 (1: 1000, ab13847, Abcam, Inc., Cambridge, UK) at 4°C overnight. Then, the sections were further incubated with secondary antibody (1: 5000, ab205718, Abcam) at 37°C for 20 min. Next, the tissues were further stained with 2, 4-diaminobutyric acid (ST033, Guangzhou Whiga Technology Co., Guangzhou, Guangdong, China), counter-stained with hematoxylin (#E607317, Sangon), and dehydrated. Thereafter, the tissues were washed and observed under a microscope (Nikon Instruments, Inc., Tokyo, Japan) with 5 fields randomly selected. Quantitative analysis for images was performed using Image J software.
ENZYME-LINKED IMMUNOSORBENT ASSAY (ELISA):
The contents of extracellular signal-regulated kinase/mitogen-activated protein kinase (ERK/MEK) and phosphorylated ERK/MEK in cells and the levels of inflammatory cytokines tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β) were detected using ELISA kits (ERK/MEK, ab176660, Abcam; TNF-α, KE10002, Proteintech Group, Inc., Wuhan, Hubei, China; IL-1β, KE10003, Proteintech) in strict accordance with the manufacturer’s instructions. In brief, 100 mg tissue homogenate was mixed with 1 mL lysis solution containing 40 μL radio-immunoprecipitation assay (RIPA) cell lysis buffer+5 μL protease inhibitor+5 μL phosphatase inhibitor+5 μL phenylmethylsulfonyl fluoride (Thermo Fisher). The mixture was then centrifuged at 15 000 rpm at 4°C for 10 min. Then, 800 μL supernatant was collected, and the protein concentration was determined using a bicinchoninic acid (BCA) method kit (Thermo Fisher Scientific Inc., Waltham, MA, USA). A total of 20 μg protein was used for ELISA. Each blank well was loaded with 100 μL sample diluent, while the residual wells were loaded with either 100 μL standard or sample for test. The ELISA plate was covered by tectorial membrane for 90 min of warm incubation at 37°C. Then, each well was loaded with 100 μL working solution of primary antibodies (prepared 15 min prior to use). Thereafter, the plate was sealed with membrane again for 1 h of incubation. Then, the liquid in wells was discarded, and each well was filled with 100 μL horseradish peroxidase (HRP)-conjugated working solution (prepared 15 min prior to use) for 30 min of warm incubation. Following color development by addition of 2,4-diaminobutyric acid, the optical density (OD) value of each well at 450 nm was determined using a microplate reader.
CELL CULTURE AND OXYGEN-GLUCOSE DEPRIVATION (OGD):
The neurogenic mouse cell line Neuro-2a (N2a, Cat. No. CCL-131) purchased from American Type Culture Collection (ATCC, Manassas, VA, USA) was cultivated in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin (all purchased from Invitrogen). Then, the cells were further incubated in a glucose-free DMEM pre-balanced in 95% N2 and 5% CO2 for oxygen-glucose deprivation to simulate an ischemic condition for 24 h. The oxygen concentration was monitored using an oxygen analyzer (Sable System, Las Vegas, USA) and maintained at less than 1%. The 293T cells used for dual-luciferase reporter gene assay were also from ATCC (Cat. No. CRL-1573). Cells were cultured in a similar condition (10% FBS-DMEM) for further use.
CONSTRUCTION OF SMALL INTERFERING (SI)-RNA TARGETING SNHG15 AND MIMIC OF MIR-18A:
The siRNA targeting SNHG15 (5′-GCCCGCUGAGCGCCUCCAGAC-3′) and the control siRNA (5′-UUCUCCGAACGUGUCACGU-3′) were purchased from GenePharma Co. (Shanghai, China) and transfected into N2a cells using Lipofectamine™ 3000 (Invitrogen, Thermo Fisher) according to the manufacturer’s instructions. The miR-18a mimic (5′-AGGGGGGGCAGGAGGGGCUCAGGG-3′) and the mimic control (Mock) (5′-UUCUCCGAAAACGUGUCACGU-3′) were also synthesized by GenePharma and transfected into N2a cells in a similar manner. Seventy hours after transfection, cells were collected for RNA and protein extraction for further experiments. In addition, cells transfected with siRNA-SNHG15 or miR-18a mimic were further treated with an ERK/MEK signaling-specific antagonist U0126 (50 nM) (#HY-12031, MedChemExpress, Monmouth Junction, NJ, USA) [20]. Twenty-four hours later, the cells were collected for further use.
5-ETHYNYL-2′-DEOXYURIDINE (EDU) LABELING ASSAY:
Well-growing passage 3 cells were collected, and the DNA replication ability of cells was detected using a cell-light EdU labeling kit (Cat. No. C10310-1, RiboBio Co., Guangzhou, Guangdong China) as per the manufacturer’s instructions. In brief, cells were seeded onto 24-well plates at 2×105 cells per well and incubated in 10% FBS-supplemented medium for 24 h. Next, the cells were treated with diluted EdU labeling reagent (1: 1000, RiboBio) for 6 h, and then the cells were fixed in 4% paraformaldehyde for 30 min and immersed in 2 μmg/mL glycine solution for 5 min. After that, the cells were incubated with 0.5% TritonX-100 (#A110694, Sangon) in phosphate-buffered saline (PBS) at room temperature for 20 min. The labeled cells were observed under a fluorescence microscope (Eclipse TS100, Nikon). Nuclei stained by 4′, 6-diamidino-2-phenylindole (DAPI) were observed by ultraviolet photoluminescence (wavelength=425 nm) while the EdU-labeled cells were observed using green photoluminescence (wavelength=590 nm), and the ratio of EdU-positive cells to total cells was evaluated by Image J software.
HOECHST 33258 STAINING:
Apoptosis of cells was determined using Hoechst 33258 staining. All procedures were performed as described in a previous report [21]. A Hoechst 33258 assay kit (Cat. No. C0003, Beyotime Biotechnology Co., Shanghai, China) was used for nuclear staining, by which the apoptotic cells were stained in bright blue, indicating pyknosis. Exponentially growing passage 3 cells were fixed in 4% paraformaldehyde for 30 min and then stained with Hoechst 33258 solution (10 μg/mL) for 5 min and photographed under an Eclipse TS100 fluorescence microscope at a 200×magnification.
FLOW CYTOMETRY:
Cell apoptosis was further measured using flow cytometry as previously described [22]. Cells were incubated with Annexin V fluorescein isothiocyanate/propidium iodide (Cat. No.556547, BD Biosciences, San Jose, CA, USA), and the proportion of apoptotic cells was evaluated using a Beckman cytometer (BD Biosciences) and FlowJo software (V11.0, BD Biosciences).
3-(4, 5-DIMETHYLTHIAZOL-2-YL)-2, 5-DIPHENYLTETRAZOLIUM BROMIDE (MTT) ASSAY:
Viability of cells was detected using an MTT assay kit (ab211091, Abcam). Briefly, 1×104 cells were seeded onto 96-well plates, and then each well was filled with 20 μL solution for 2 h. Next, the OD value at 590 nm was evaluated using a spectrophotometer.
REVERSE TRANSCRIPTION QUANTITATIVE POLYMERASE CHAIN REACTION (RT-QPCR):
An RNA purification kit (Cat. No. Dx17200, Norgen Biotek, Thorold, Canada) was applied to separate cytoplastic and nuclear RNA. RT-qPCR was performed using a SYBR Green qPCR Mix kit (Thermo Fisher) in strict accordance with the kit’s instructions on an ABI real-time qPCR system (Thermo Fisher). The primer sequences are shown in Table 1.
:
An RNA FISH assay was performed in line with the instructions of the kit (Gene Pharma). In detail, the N2a cells were seeded into 48-well plates, washed in PBS, and then fixed in 4% paraformaldehyde for 15 min. Next, the cells were incubated with 0.1% Triton X-100 for 15 min and hybridized with fluorescence-coupled G043225 probe in the dark overnight. Next, the compounds were washed in 0.1% Tween 20 (#CAS: 9005-64-5) for 5 min and then washed with formamide: 2×citrate sodium (SSC; 50: 50) for 5 min, while the nuclei were stained by DAPI. Representative images were obtained using an Eclipse TS100 fluorescence microscope at 200× magnification.
DUAL-LUCIFERASE REPORTER GENE ASSAY:
The binding relationships between miR-18a and SNHG15 and between miR-18a and CXCL13 were first predicted in StarBase (http://starbase.sysu.edu.cn/) and then validated by a dual-luciferase reporter gene assay, as previously reported [23]. The luciferase vectors containing the binding sequences and the pRL-TK Renilla luciferase vector (internal reference) were co-transfected with miR-18a mimic or NC mimic into 293T cells for 36 h. Then, the cell lysates were collected, and the relative luciferase activity was detected on a Dual-luciferase Reporter Gene System (Cat. No. RG089S, Beyotime) as per the manufacturer’s instructions.
WESTERN BLOT ANALYSIS:
Phosphorylation of ERK1/2 and MEK1/2 were further validated by Western blot analysis. Phospho-p44/42 MAPK (ERK1/2) (Thr202/Tyr204) (Cat. No. 4320, 1: 1000) and Phospho-MEK1/2 (Ser221) (Car: 2338, 1: 1000) were purchased from Cell Signaling Technology, Inc. (Beverly, MA, USA). The glyceraldehyde-3-phosphate dehydrogenase (ab8245, 1: 5000) antibody was from Abcam, and the HRP-conjugated secondary antibody (Cat. No. SA00001-2, 1: 5000) was from Proteintech. Protein lysates were prepared using RIPA lysis buffer and the concentration was determined using the BCA kit. Then, 25 μg total protein was collected and separated by 8% sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred onto polyvinylidene fluoride membranes (EMD Millipore Corp., Billerica, MA, USA). Next, the membranes were blocked using skimmed milk at room temperature for 30 min, washed, and incubated with the primary antibodies at 4°C overnight, and then with the secondary antibody at room temperature for 1 h. The blots were developed using the enhanced chemiluminescence Western blot substrate (Thermo Fisher). Relative protein expression was determined using Image J software.
STATISTICAL ANALYSIS:
Data were analyzed using SPSS 21.0 (IBM Corp. Armonk, NY, USA). Kolmogorov-Smirnov testing was performed to determine normal distribution of data. Measurement data are exhibited as mean±standard deviation (SD). Differences between 2 groups were evaluated using the
Results
ALTERED GENE EXPRESSION IN MCAO-INDUCED MOUSE MODEL:
A MCAO-induced mouse model was established to evaluate the role of SNHG15 in ischemic stroke. The TTC staining results showed that the stained area in mouse brain tissues was notably reduced following MCAO, indicating a significant increase in infarct size (Figure 1A). In addition, the scoring of neurologic deficit in mice based on the method by Longa et al. suggested that the mice showed moderate to severe neurological dysfunction following MCAO (Figure 1B). These results suggest successful establishment of a mouse model of ischemic stroke. SNHG15 expression was notably increased, while miR-18a expression was decreased, and CXCL13 expression was increased following MCAO procedures (Figure 1C). The immunohistochemical staining results suggested that caspase-3 expression was significantly increased (Figure 1D). We also examined the levels of pro-inflammatory cytokines TNF-α and IL-1β in mouse brain tissues, and found the production of these cytokines was notably increased following MCAO (Figure 1E). The ELISA results also showed that activation of the ERK/MEK signaling pathway was inhibited after MCAO (Figure 1F).
SILENCING OF SNHG15 ATTENUATES N2A CELL APOPTOSIS AFTER OGD TREATMENT:
The OGD procedure was performed on N2a cell lines to simulate an ischemic stroke condition at the cell level. It was found that expression of SNHG15 and CXCL13 was increased while miR-18a expression was decreased in OGD-treated N2a cells. Next, siRNA targeting SNHG15 was introduced in cells, after which SNHG15 and CXCL13 expression was significantly reduced and miR-18a expression was increased (Figure 2A). OGD treatment inhibited viability and promoted apoptosis of N2a cells, while silencing of SNHG15 reversed these changes (Figure 2B–2E).
SNHG15 SERVES AS A CERNA FOR MIR-18A TO MEDIATE CXCL13 EXPRESSION:
RT-qPCR was performed to identify SNHG15 expression in nuclear and cytoplastic RNA. It was found that SNHG15 was mainly sub-localized in cytoplasm (Figure 3A). A FISH assay was further performed to validate the subcellular localization of SNHG15 in N2a cells, which identified the cytoplasm-localization of the red SNHG15-probes (Figure 3B), indicating SNHG15 exerts functions through the ceRNA network. The binding relationships between miR-18a and SNHG15 and between miR-18a and the 3′-UTR of CXCL13 mRNA were predicted in StarBase (http://starbase.sysu.edu.cn/) and validated through dual-luciferase reporter gene assays. The luciferase activities in cells co-transfected with SNHG15-WT and miR-18a mimic and in cells co-transfected with CXCL13-WT and miR-18a mimic were notably decreased, while the activity in cells subjected to other co-transfection showed no significant changes (Figure 3C, 3D). Moreover, the ELISA results found that OGD treatment inactivated the ERK/MEK signaling pathway, while the phosphorylation of ERK and MEK was significantly increased following SNHG15 silencing (Figure 3E). A similar trend was shown by Western blot analysis, where the phosphorylation of ERK1/2 and MEK1/2 was reduced after OGD treatment but increased following SNHG15 silencing (Figure 3F).
OVEREXPRESSION OF MIR-18A INHIBITS APOPTOSIS AND PROMOTES VIABILITY OF N2A CELLS:
Artificial upregulation of miR-18a was introduced in OGD-treated N2a cells by transfection of miR-18a mimic, after which miR-18a expression was significantly increased according to RT-qPCR (Figure 4A). Next, the EdU labeling assay found that overexpression of miR-18a promoted viability of OGD-treated N2a cells (Figure 4B). Moreover, miR-18a mimic transfection led to significantly reduced apoptosis in N2a cells according to Hoechst staining and flow cytometry (Figure 4C, 4D).
THE ERK/MEK ANTAGONIST REVERSES THE ROLES OF SNHG15 SIRNA AND MIR-18A MIMIC IN N2A CELLS:
To further identify the involvement of the ERK/MEK signaling pathway in silenced SNHG15/overexpressed miR-18a-mediated events, an ERK/MEK antagonist, U0126 (50 nM), was further introduced into N2a cells in the presence of SNHG15 silencing or miR-18a overexpression. It was found that ERK/MEK defect led to decreased cell viability and increased cell apoptosis in N2a cells with silenced SNHG15 or overexpressed miR-18a (Figure 5A–5C).
Discussion
Ischemic stroke accounts for approximately 60% of all cerebrovascular disorders, followed by cerebral hemorrhage and subarachnoid hemorrhage [4]. Aberrant lncRNA expression has been validated to be closely linked with cell death in ischemic stroke progression [5]. SNHG15 has been widely identified as an onco-lncRNA in multiple cancer types [12], but its role in other diseases is less studied. The present study validated that SNHG15 is highly expressed in MCAO-treated mice and OGD-treated N2a cells, which aggravates neuronal injury after ischemic stroke with the involvement of the miR-18a/CXCL13/ERK/MEK axis.
Firstly, a mouse model of ischemic stroke was induced by MCAO, a common method for stoke induction in rodents [24]. We found SNHG15 expression was notably increased in MCAO-treated mice compared to the sham-operated ones. The same trend was found in OGD-treated N2a cells. Many lncRNAs have been identified as having critical influences in stroke progression. For instance, lncRNA H19 was found to trigger neuroinflammation following ischemic stroke by triggering microglial M1 polarization [25]. Likewise, lncRNA MEG3 was found to function as a cell death promoter in ischemia by interacting with p53 to mediate ischemic damage [26]. On the other hand, some lncRNAs such as lncRNA HOTTIP [27] and lncRNA-N1LR [28] have been suggested to have neuroprotective roles after ischemic stroke through different mechanisms. SNHG15 has recently been identified as an oncogene in many human cancer types [29–31]. In addition to this, intriguingly, SNHG15 was found to be highly expressed in patients with ischemic stroke and to be positively correlated with the severity [14]. The present study corroborated this finding, and based on this result, we further confirmed that artificial silencing of SNHG15 led to a decrease in apoptosis but an increase in viability of OGD-treated N2a cells, indicating SNHG15 might play a crucial role in neuronal death following ischemic stroke. SNHG15 silencing was found to inhibit caspase-3 expression in cells, which is an important mediator of apoptosis [32]. This result further validated the pro-apoptotic role of SNHG15 from the molecular perspective. In addition, neuroinflammation is a major process occurring during ischemic stroke progression, which promotes further injury and cell death, thus serving as a promising target for ischemic stroke treatment [33,34]. The present study identified that silencing of SNHG15 led to decreased production of major pro-inflammatory cytokines (TNF-α and IL-1β) in the brain tissues of MCAO-induced mice, further demonstrating the neuroprotective role of SNHG15 silencing.
The above findings led us to further explore the mechanisms involved. We identified miR-18a as a target of SNHG15, which could further bind to CXCL13 mRNA. miRNAs are well known to play diverse functions in ischemic stroke progression and inflammatory responses, with a wide array of miRNAs found to be closely linked to the development of this disease [35,36]. miR-18a is a member of the miR-17-92 miRNA cluster, whose close association with the neurogenesis of CNS has been revealed [37]. A member of this family, miR-17, was found to be expressed at a 9.9-fold higher level in patients with ischemic stroke than in healthy individuals [38], while another important member, miR-19a, was found to be underexpressed in such patients [39]. In this paper, miR-18a was found to be underexpressed in MCAO-induced mice and in OGD-treated N2a cells, while further silencing of SNHG15 led to increased miR-18a expression. In addition, upregulation of miR-18a by miRNA mimic in this study was found to increase viability, but it decreased apoptosis of OGD-treated N2a cells. This is in line with a previous study reporting that silencing of miR-18a could trigger death and retard proliferation of neural progenitor cells after stroke [40]. In addition, we found that CXCL13 was a target of miR-18a, and the expression profile of CXCL13 was positively correlated with SNHG15. CXCL13 is a chemokine that recruits B cells during inflammation and is elevated in the CNS by infiltrating immune cells at many sclerosis lesion sites and during CNS infection [41]. Upregulation of CXCL13 is found following traumatic brain injury [42] and ischemic stroke [43], leading to aggravated inflammation. In addition, our study found that the ERK/MEK signaling pathway is inactivated in MCAO-treated mice and OGD-treated N2a cells, while silencing of SNHG15 activated this signaling pathway in N2a cells, as shown by ELISA and Western blot analysis results. ERK (ERK1 and ERK2) is activated upon MEK (MEK1 and MEK2) phosphorylation, and its aberrant expression has been suggested to promote cancer cell proliferation and inhibit cell apoptosis [44]. The pro-proliferative and anti-apoptotic roles of ERK/MEK were substantiated in N2a cells [45]. Activation of the ERK/MEK pathway can be regarded as a neuroprotection biomarker following ischemic injury [46]. Moreover, N2a cells with stable silencing of SNHG15 or overexpression of miR-18a were further introduced with an ERK antagonist, U0126, after which the neuroprotective roles of SNHG15-siRNA and miR-18a in N2a cells were notably antagonized. This finding further identified the involvement of the ERK/MEK signaling in the SNHG15-mediated events. In addition to the ERK/MEK signaling, some other signaling pathways such as Notch [47], Wnt [48], and TGF/Smad [49] are noted to be closely linked to cell death and pathogenesis of ischemic stroke. Whether these signaling pathways are affected by the molecules mentioned in this study is unknown and warrants further research. We plan to explore the involvement of these potential pathways in our further investigations.
Conclusions
We identified a novel ceRNA network involving SNHG15/miR-18a/CXCL13 that can influence cell apoptosis and proliferation, as well as inflammation, following ischemic stroke. Silencing of SNHG15 or overexpression of miR-18a can elevate viability and reduce apoptosis of OGD-treated N2a cells, during which the ERK/MEK signaling activation may be involved. These findings may provide novel insights into ischemic treatment, suggesting that SNHG15 is a potential therapeutic target. However, the molecular mechanisms involved are too complex to be fully identified. Here, we just identified a single ceRNA network. Given the present study’s limitations, we plan to perform NextGen sequencing to provide a more holistic picture of this network in our future studies. We would also like to discover more details of the independent roles of CXCL13 and its correlation with the ERK/MEK signaling pathway, as well as the other potential signaling pathways involved. We also hope more research will be launched in this field to validate our findings and to offer more ideas for ischemic stroke treatment.
Figures
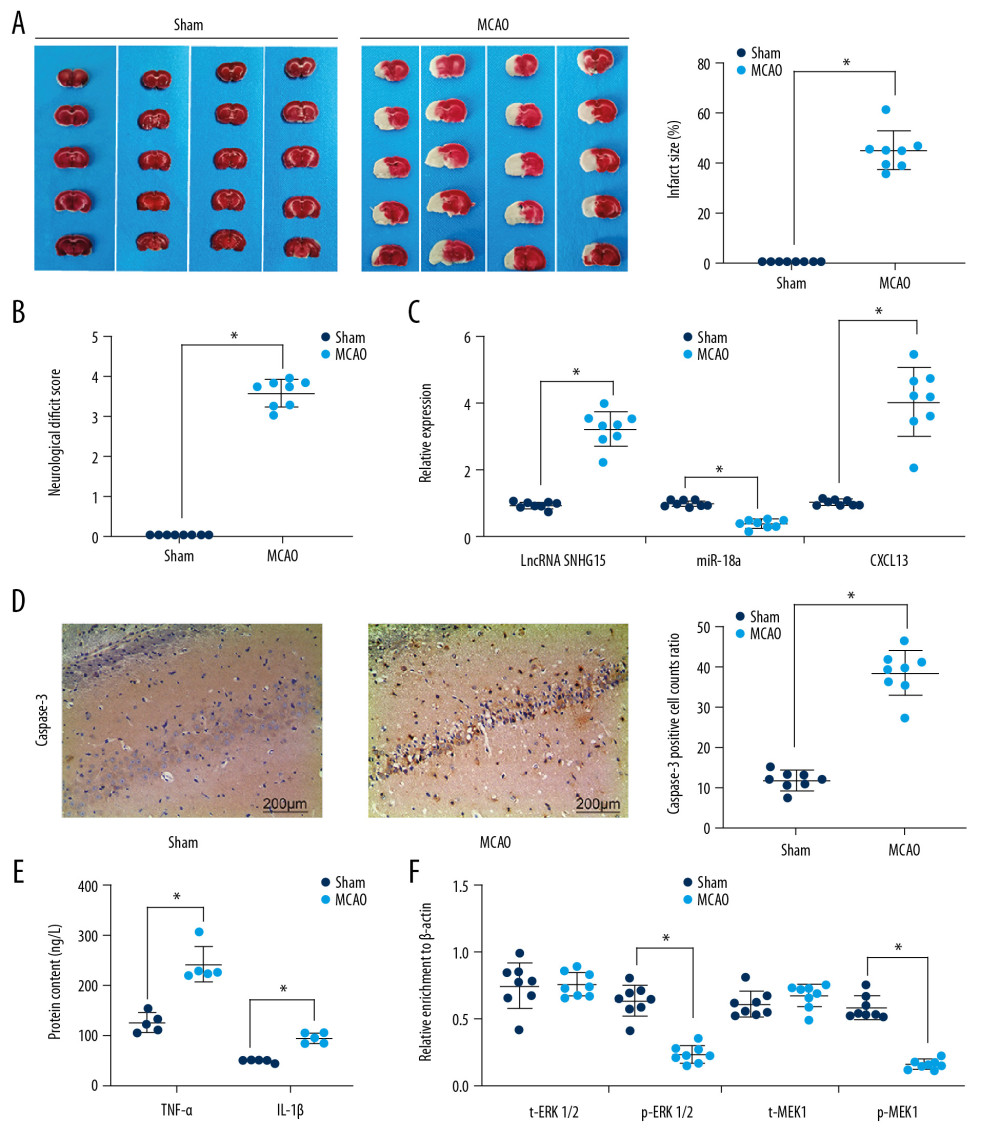 Figure 1. Altered gene expression in mice following MCAO procedures. Model mice were subjected to MCAO with 2 h of ischemia and 24 h of reperfusion, while sham-operated mice underwent the same procedures without ischemia, and then the brains of all mice were collected for the following experiments. (A) Representative images of mouse brain tissue sections after TTC staining. (B) Evaluation of the neurological deficit scores. (C) Expression of SNHG15, miR-18a, and CXCL13 in mouse brain tissues was detected using RT-qPCR. (D) Caspase-3 expression in mouse hippocampal tissues was evaluated using immunohistochemistry staining. (E) Protein levels of pro-inflammatory cytokines TNF-α and IL-1β in mouse brain were determined using ELISA kits. (F) Contents of total MEK/MRK and phosphorylated MEK/MRK in mouse brain tissues was detected using ELISA kits. Data are shown as mean±SD based on 3 independent experiments. N=8, * p<0.05. In panels A, B, and D, data were analyzed using the unpaired t test, while data in panels C, E, and F were analyzed using one-way ANOVA.
Figure 1. Altered gene expression in mice following MCAO procedures. Model mice were subjected to MCAO with 2 h of ischemia and 24 h of reperfusion, while sham-operated mice underwent the same procedures without ischemia, and then the brains of all mice were collected for the following experiments. (A) Representative images of mouse brain tissue sections after TTC staining. (B) Evaluation of the neurological deficit scores. (C) Expression of SNHG15, miR-18a, and CXCL13 in mouse brain tissues was detected using RT-qPCR. (D) Caspase-3 expression in mouse hippocampal tissues was evaluated using immunohistochemistry staining. (E) Protein levels of pro-inflammatory cytokines TNF-α and IL-1β in mouse brain were determined using ELISA kits. (F) Contents of total MEK/MRK and phosphorylated MEK/MRK in mouse brain tissues was detected using ELISA kits. Data are shown as mean±SD based on 3 independent experiments. N=8, * p<0.05. In panels A, B, and D, data were analyzed using the unpaired t test, while data in panels C, E, and F were analyzed using one-way ANOVA. 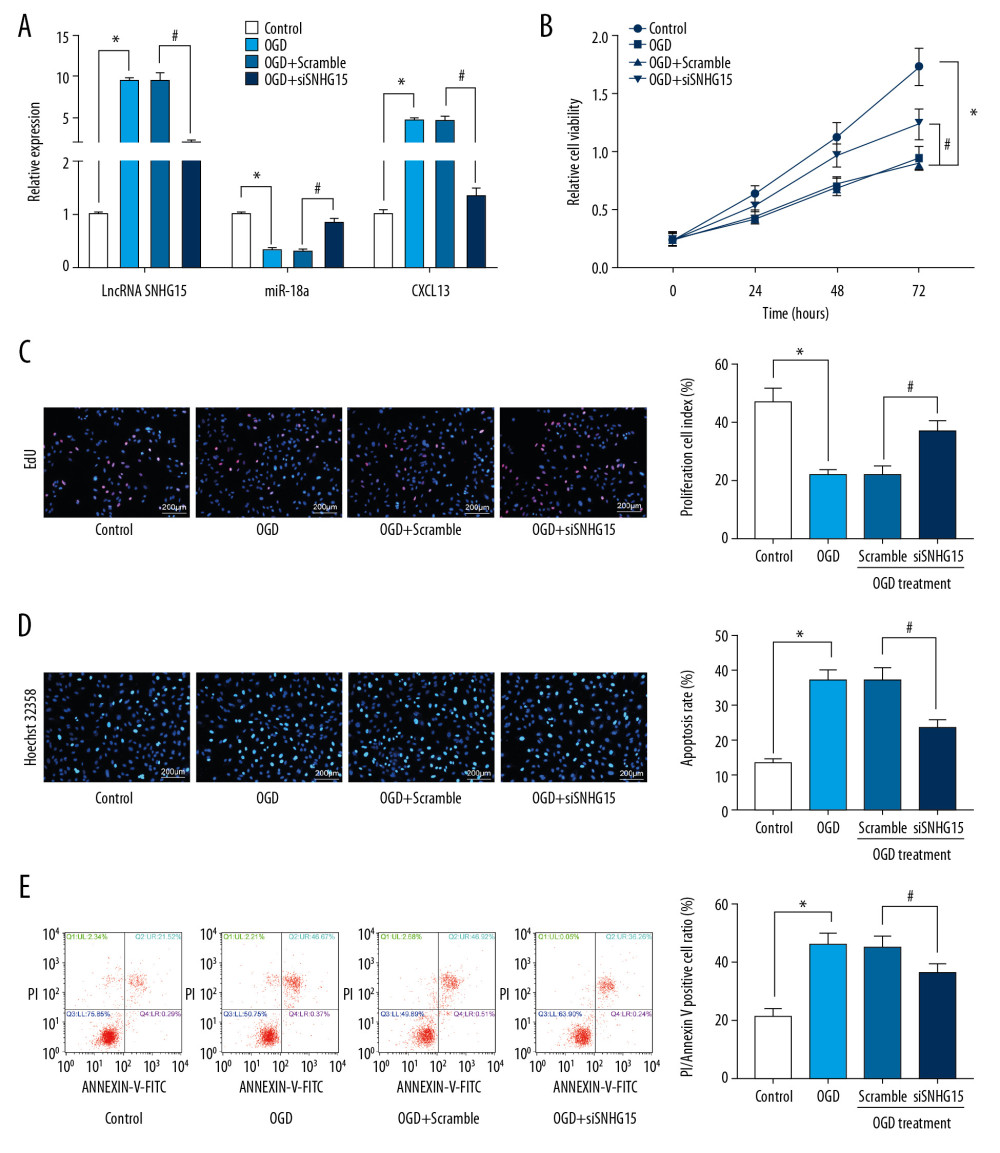 Figure 2. Silencing of SNHG15 inhibits OGD-induced N2a cell apoptosis. N2a cells were subjected to OGD treatment and collected for experiments. (A) Expression of SNHG15, miR-18a, and CXCL13 in cells before and after si-SNHG1 treatment was detected using RT-qPCR. (B, C) Viability of N2a cells was detected using MTT and EdU labeling assays. (D, E) Apoptosis of N2a cells was detected using Hoechst 33258 staining and flow cytometry. Data were shown as mean±SD based on 3 independent experiments. * p<0.05. In panels A, C–E, data were analyzed using one-way ANOVA while data in panel B were analyzed using two-way ANOVA, and Tukey’s multiple comparison test was used for post hoc analysis.
Figure 2. Silencing of SNHG15 inhibits OGD-induced N2a cell apoptosis. N2a cells were subjected to OGD treatment and collected for experiments. (A) Expression of SNHG15, miR-18a, and CXCL13 in cells before and after si-SNHG1 treatment was detected using RT-qPCR. (B, C) Viability of N2a cells was detected using MTT and EdU labeling assays. (D, E) Apoptosis of N2a cells was detected using Hoechst 33258 staining and flow cytometry. Data were shown as mean±SD based on 3 independent experiments. * p<0.05. In panels A, C–E, data were analyzed using one-way ANOVA while data in panel B were analyzed using two-way ANOVA, and Tukey’s multiple comparison test was used for post hoc analysis. 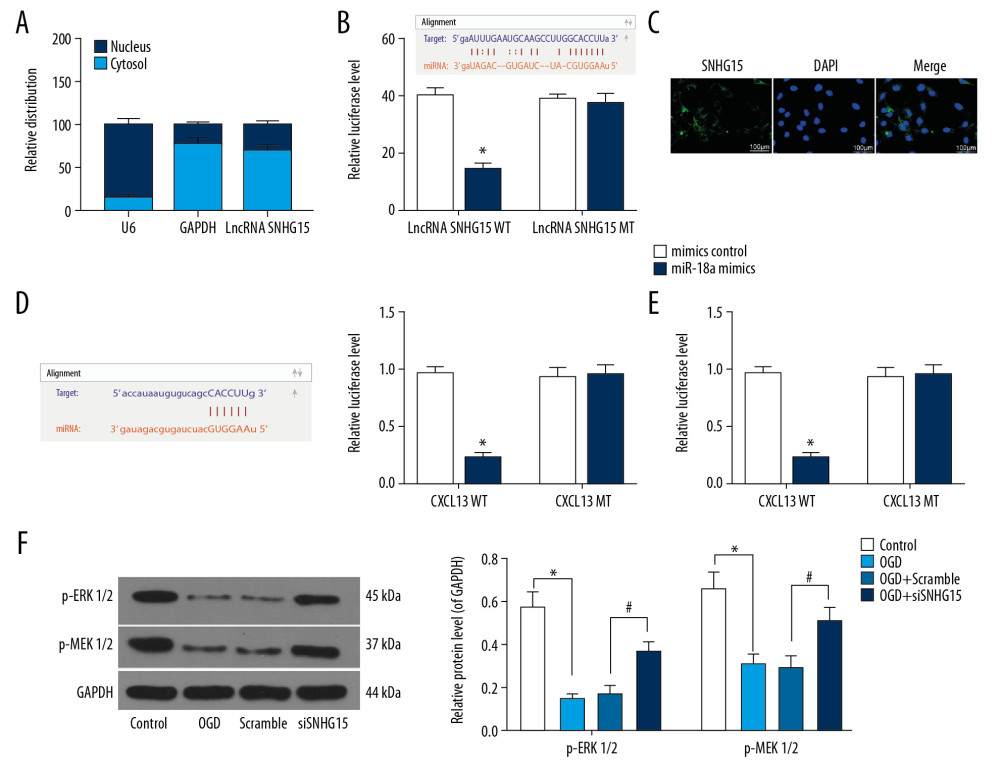 Figure 3. SNHG15 serves as a ceRNA for miR-18a to mediate THBS2 expression. (A, B) Sub-cellular localization of SNHG15 in N2a cells was determined by a RT-qPCR (A) and a FISH assay (B). (C, D) Binding relationships between miR-18a and SNHG15 (C), and between miR-18a and the 3′-UTR of CXCL13 mRNA (D) were predicted on Starbase (http://starbase.sysu.edu.cn/) and validated through dual-luciferase reporter gene assay. (E) Contents of total ERK/MEK and phosphorylated ERK/MEK in N2a cells was quantified by ELISA kits. F, phosphorylation of ERK1/2 and MEK1/2 was determined by Western blot analysis. Data were shown as mean±SD based on 3 independent experiments. * p<0.05. In panels A and F, data were analyzed using two-way ANOVA, while data in panels C–E were analyzed using one-way ANOVA, and Tukey’s multiple comparison test was used for post hoc analysis.
Figure 3. SNHG15 serves as a ceRNA for miR-18a to mediate THBS2 expression. (A, B) Sub-cellular localization of SNHG15 in N2a cells was determined by a RT-qPCR (A) and a FISH assay (B). (C, D) Binding relationships between miR-18a and SNHG15 (C), and between miR-18a and the 3′-UTR of CXCL13 mRNA (D) were predicted on Starbase (http://starbase.sysu.edu.cn/) and validated through dual-luciferase reporter gene assay. (E) Contents of total ERK/MEK and phosphorylated ERK/MEK in N2a cells was quantified by ELISA kits. F, phosphorylation of ERK1/2 and MEK1/2 was determined by Western blot analysis. Data were shown as mean±SD based on 3 independent experiments. * p<0.05. In panels A and F, data were analyzed using two-way ANOVA, while data in panels C–E were analyzed using one-way ANOVA, and Tukey’s multiple comparison test was used for post hoc analysis. 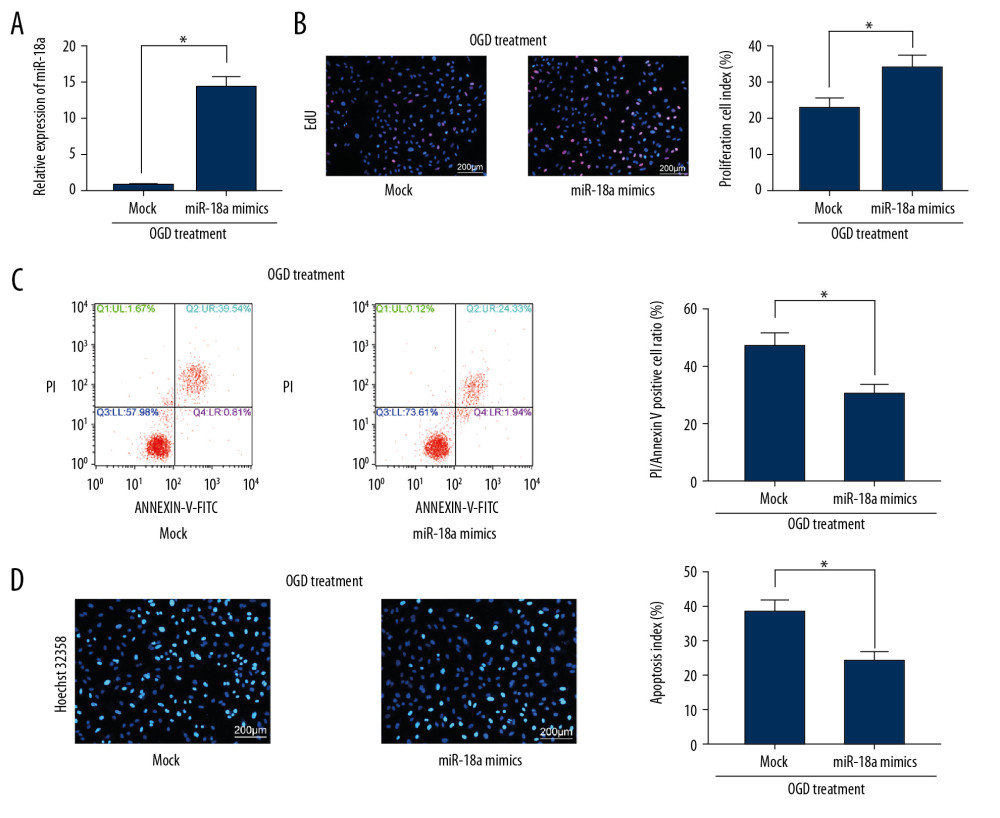 Figure 4. Overexpression of miR-18a inhibits apoptosis and promotes viability of N2a cells. (A) MiR-18a expression before and after miR-18a mimic transfection was performed using RT-qPCR. (B) Viability of OGD-treated N2a cells was detected using EdU labeling assay. (C, D) Apoptosis of OGD-treated N2a cells was detected using Hoechst 33258 staining (C) and flow cytometry (D). Data are shown as mean±SD based on 3 independent experiments. * p<0.05. Data were analyzed using the unpaired t test.
Figure 4. Overexpression of miR-18a inhibits apoptosis and promotes viability of N2a cells. (A) MiR-18a expression before and after miR-18a mimic transfection was performed using RT-qPCR. (B) Viability of OGD-treated N2a cells was detected using EdU labeling assay. (C, D) Apoptosis of OGD-treated N2a cells was detected using Hoechst 33258 staining (C) and flow cytometry (D). Data are shown as mean±SD based on 3 independent experiments. * p<0.05. Data were analyzed using the unpaired t test. 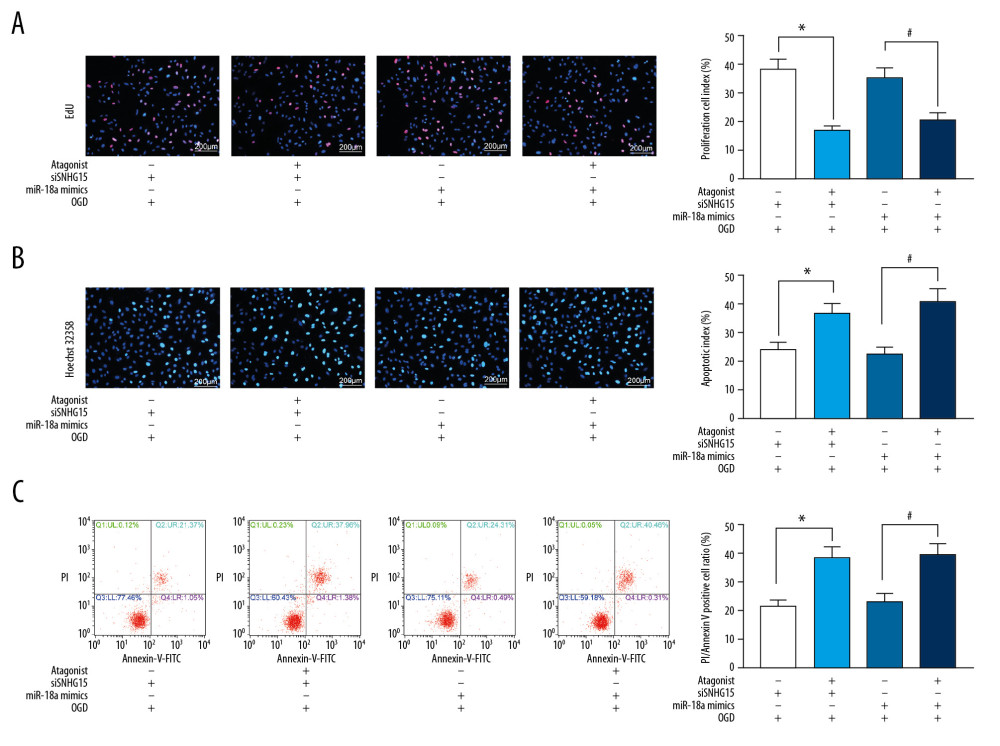 Figure 5. The ERK/MEK antagonist reverses the roles of SNHG15 siRNA and miR-18a mimic in N2a cells. An ERK/MEK antagonist, U0126, was introduced into N2a cells with silenced SNHG15 or overexpressed miR-18a. (A) Viability of N2a cells was detected using EdU labeling assay. (B, C) Apoptosis of OGD-treated N2a cells was detected using Hoechst 33258 staining (B) and flow cytometry (C), respectively. Data are shown as mean±SD based on 3 independent experiments. * p<0.05. Data were analyzed using one-way ANOVA.
Figure 5. The ERK/MEK antagonist reverses the roles of SNHG15 siRNA and miR-18a mimic in N2a cells. An ERK/MEK antagonist, U0126, was introduced into N2a cells with silenced SNHG15 or overexpressed miR-18a. (A) Viability of N2a cells was detected using EdU labeling assay. (B, C) Apoptosis of OGD-treated N2a cells was detected using Hoechst 33258 staining (B) and flow cytometry (C), respectively. Data are shown as mean±SD based on 3 independent experiments. * p<0.05. Data were analyzed using one-way ANOVA. 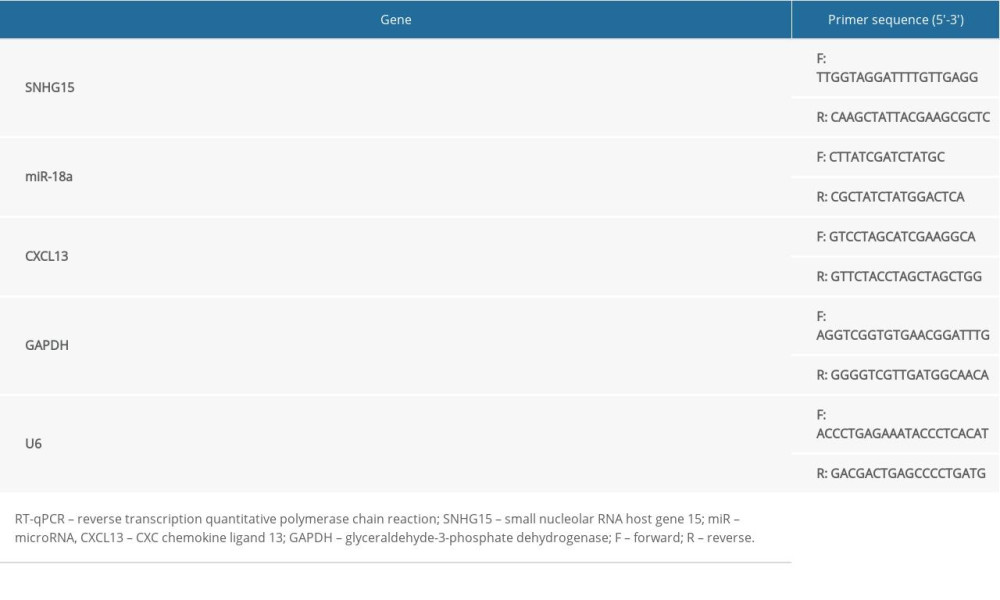
References
1. Yang J, Chen M, Cao RY, The role of circular RNAs in cerebral ischemic diseases: Ischemic stroke and cerebral ischemia/reperfusion injury: Adv Exp Med Biol, 2018; 1087; 309-25
2. Veno SK, Schmidt EB, Bork CS, Polyunsaturated fatty acids and risk of ischemic stroke: Nutrients, 2019; 11(7); 1467
3. Radak D, Katsiki N, Resanovic I, Apoptosis and acute brain ischemia in ischemic stroke: Curr Vasc Pharmacol, 2017; 15(2); 115-22
4. Fukuta T, Ishii T, Asai T, Oku N, Applications of liposomal drug delivery systems to develop neuroprotective agents for the treatment of ischemic stroke: Biol Pharm Bull, 2019; 42(3); 319-26
5. Alishahi M, Ghaedrahmati F, Kolagar TA, Long non-coding RNAs and cell death following ischemic stroke: Metab Brain Dis, 2019; 34(5); 1243-51
6. Dai X, Zhang S, Zaleta-Rivera K, RNA: interactions drive functionalities: Mol Biol Rep, 2020; 47(2); 1413-34
7. Xue M, Zhuo Y, Shan B, MicroRNAs, long noncoding RNAs, and their functions in human disease: Methods Mol Biol, 2017; 1617; 1-25
8. Stanzione R, Cotugno M, Bianchi F, Pathogenesis of ischemic stroke: Role of epigenetic mechanisms: Genes (Basel), 2020; 11(1); 89
9. Smillie CL, Sirey T, Ponting CP, Complexities of post-transcriptional regulation and the modeling of ceRNA crosstalk: Crit Rev Biochem Mol Biol, 2018; 53(3); 231-45
10. Zhou XB, Lai LF, Xie GB, LncRNAGAS5 sponges miRNA-221 to promote neurons apoptosis by up-regulated PUMA under hypoxia condition: Neurol Res, 2020; 42(1); 8-16
11. Deng Y, Chen D, Gao F, Silencing of long non-coding RNA GAS5 suppresses neuron cell apoptosis and nerve injury in ischemic stroke through inhibiting DNMT3B-dependent MAP4K4 methylation: Transl Stroke Res, 2020 [Online ahead of print]
12. Shuai Y, Ma Z, Lu J, Feng J, LncRNA SNHG15: A new budding star in human cancers: Cell Prolif, 2020; 53(1); e12716
13. Li Z, Zhang J, Zheng H, Modulating lncRNA SNHG15/CDK6/miR-627 circuit by palbociclib, overcomes temozolomide resistance and reduces M2-polarization of glioma associated microglia in glioblastoma multiforme: J Exp Clin Cancer Res, 2019; 38(1); 380
14. Deng QW, Li S, Wang H, Differential long noncoding RNA expressions in peripheral blood mononuclear cells for detection of acute ischemic stroke: Clin Sci (Lond), 2018; 132(14); 1597-614
15. Huang H, Wei G, Wang C, A functional polymorphism in the promoter of miR-17-92 cluster is associated with decreased risk of ischemic stroke: BMC Med Genomics, 2019; 12(1); 159
16. Ferreira R, Santos T, Amar A, MicroRNA-18a improves human cerebral arteriovenous malformation endothelial cell function: Stroke, 2014; 45(1); 293-97
17. Huber AK, Irani DN, Targeting CXCL13 during neuroinflammation: Adv Neuroimmune Biol, 2015; 6(1); 1-8
18. Yin XM, Luo Y, Cao G, Bid-mediated mitochondrial pathway is critical to ischemic neuronal apoptosis and focal cerebral ischemia: J Biol Chem, 2002; 277(44); 42074-81
19. Longa EZ, Weinstein PR, Carlson S, Cummins R, Reversible middle cerebral artery occlusion without craniectomy in rats: Stroke, 1989; 20(1); 84-91
20. Bessard A, Fremin C, Ezan F: Oncogene, 2008; 27(40); 5315-25
21. Bronisz-Kowalczyk A, Buzanska L, Kowalczyk D, Domanska-Janik K, Extracellular signal-regulated kinase suppression is not involved in apoptosis of neuroblastoma N2a cells induced by protein kinase C inhibition: Folia Neuropathol, 2000; 38(1); 13-21
22. Wang R, Ma Z, Feng L, LncRNA MIR31HG targets HIF1A and P21 to facilitate head and neck cancer cell proliferation and tumorigenesis by promoting cell-cycle progression: Mol Cancer, 2018; 17(1); 162
23. Ma H, Jin S, Yang W, Interferon-alpha enhances the antitumour activity of EGFR-targeted therapies by upregulating RIG-I in head and neck squamous cell carcinoma: Br J Cancer, 2018; 118(4); 509-21
24. Liu F, McCullough LD, Middle cerebral artery occlusion model in rodents: methods and potential pitfalls: J Biomed Biotechnol, 2011; 2011 464701
25. Wang J, Zhao H, Fan Z, Long noncoding RNA H19 promotes neuroinflammation in ischemic stroke by driving histone deacetylase 1-dependent M1 microglial polarization: Stroke, 2017; 48(8); 2211-21
26. Yan H, Yuan J, Gao L, Long noncoding RNA MEG3 activation of p53 mediates ischemic neuronal death in stroke: Neuroscience, 2016; 337; 191-99
27. Wang Y, Li G, Zhao L, Lv J, Long noncoding RNA HOTTIP alleviates oxygen-glucose deprivation-induced neuronal injury via modulating miR-143/hexokinase 2 pathway: J Cell Biochem, 2018; 119(12); 10107-17
28. Wu Z, Wu P, Zuo X, LncRNA-N1LR enhances neuroprotection against ischemic stroke probably by inhibiting p53 phosphorylation: Mol Neurobiol, 2017; 54(10); 7670-85
29. Qu C, Dai C, Guo Y, Long noncoding RNA SNHG15 serves as an oncogene and predicts poor prognosis in epithelial ovarian cancer: Onco Targets Ther, 2019; 12; 101-11
30. Tong J, Ma X, Yu H, Yang J, SNHG15: a promising cancer-related long noncoding RNA: Cancer Manag Res, 2019; 11; 5961-69
31. Liu LB, Jiang ZJ, Jiang XL, Wang S, Up-regulation of SNHG15 facilitates cell proliferation, migration, invasion and suppresses cell apoptosis in breast cancer by regulating miR-411-5p/VASP axis: Eur Rev Med Pharmacol Sci, 2020; 24(4); 1899-912
32. Lossi L, Castagna C, Merighi A, Caspase-3 mediated cell death in the normal development of the mammalian cerebellum: Int J Mol Sci, 2018; 19(12); 3999
33. Dabrowska S, Andrzejewska A, Lukomska B, Janowski M, Neuroinflammation as a target for treatment of stroke using mesenchymal stem cells and extracellular vesicles: J Neuroinflammation, 2019; 16(1); 178
34. Jayaraj RL, Azimullah S, Beiram R, Neuroinflammation: friend and foe for ischemic stroke: J Neuroinflammation, 2019; 16(1); 142
35. Li G, Morris-Blanco KC, Lopez MS, Impact of microRNAs on ischemic stroke: From pre- to post-disease: Prog Neurobiol, 2018; 163–64; 59-78
36. Khoshnam SE, Winlow W, Farzaneh M, The interplay of microRNAs in the inflammatory mechanisms following ischemic stroke: J Neuropathol Exp Neurol, 2017; 76(7); 548-61
37. Taylor SM, Giuffre E, Moseley P, Hitchcock PF, The MicroRNA, miR-18a, regulates NeuroD and photoreceptor differentiation in the retina of zebrafish: Dev Neurobiol, 2019; 79(2); 202-19
38. Wu J, Du K, Lu X, Elevated expressions of serum miR-15a, miR-16, and miR-17-5p are associated with acute ischemic stroke: Int J Clin Exp Med, 2015; 8(11); 21071-79
39. Jickling GC, Ander BP, Zhan X, microRNA expression in peripheral blood cells following acute ischemic stroke and their predicted gene targets: PLoS One, 2014; 9(6); e99283
40. Liu XS, Chopp M, Wang XL, MicroRNA-17-92 cluster mediates the proliferation and survival of neural progenitor cells after stroke: J Biol Chem, 2013; 288(18); 12478-88
41. Monson NL, Ortega SB, Ireland SJ, Repetitive hypoxic preconditioning induces an immunosuppressed B cell phenotype during endogenous protection from stroke: J Neuroinflammation, 2014; 11; 22
42. Huang C, Sakry D, Menzel L, Lack of NG2 exacerbates neurological outcome and modulates glial responses after traumatic brain injury: Glia, 2016; 64(4); 507-23
43. Chapman KZ, Ge R, Monni E, Inflammation without neuronal death triggers striatal neurogenesis comparable to stroke: Neurobiol Dis, 2015; 83; 1-15
44. Kulshrestha A, Katara GK, Ibrahim SA, Targeting V-ATPase isoform restores cisplatin activity in resistant ovarian cancer: Inhibition of autophagy, endosome function, and ERK/MEK Pathway: J Oncol, 2019; 2019 2343876
45. Zhong W, Li YC, Huang QY, Tang XQ, lncRNA ANRIL ameliorates oxygen and glucose deprivation (OGD) induced injury in neuron cells via miR-199a-5p/CAV-1 Axis, 2020; 45(4); 772-82
46. Feng HX, Li CP, Shu SJ: Acta Pharmacol Sin, 2019; 40(2); 160-69
47. Arumugam TV, Baik SH, Balaganapathy P, Notch signaling and neuronal death in stroke: Prog Neurobiol, 2018; 165–67; 103-16
48. Scott EL, Brann DW, Estrogen regulation of Dkk1 and Wnt/beta-catenin signaling in neurodegenerative disease: Brain Res, 2013; 1514; 63-74
49. Zhu H, Gui Q, Hui X, TGF-beta1/Smad3 signaling pathway suppresses cell apoptosis in cerebral ischemic stroke rats: Med Sci Monit, 2017; 23; 366-76
Errate

Figures
Figure 1. Altered gene expression in mice following MCAO procedures. Model mice were subjected to MCAO with 2 h of ischemia and 24 h of reperfusion, while sham-operated mice underwent the same procedures without ischemia, and then the brains of all mice were collected for the following experiments. (A) Representative images of mouse brain tissue sections after TTC staining. (B) Evaluation of the neurological deficit scores. (C) Expression of SNHG15, miR-18a, and CXCL13 in mouse brain tissues was detected using RT-qPCR. (D) Caspase-3 expression in mouse hippocampal tissues was evaluated using immunohistochemistry staining. (E) Protein levels of pro-inflammatory cytokines TNF-α and IL-1β in mouse brain were determined using ELISA kits. (F) Contents of total MEK/MRK and phosphorylated MEK/MRK in mouse brain tissues was detected using ELISA kits. Data are shown as mean±SD based on 3 independent experiments. N=8, * p<0.05. In panels A, B, and D, data were analyzed using the unpaired t test, while data in panels C, E, and F were analyzed using one-way ANOVA.Figure 2. Silencing of SNHG15 inhibits OGD-induced N2a cell apoptosis. N2a cells were subjected to OGD treatment and collected for experiments. (A) Expression of SNHG15, miR-18a, and CXCL13 in cells before and after si-SNHG1 treatment was detected using RT-qPCR. (B, C) Viability of N2a cells was detected using MTT and EdU labeling assays. (D, E) Apoptosis of N2a cells was detected using Hoechst 33258 staining and flow cytometry. Data were shown as mean±SD based on 3 independent experiments. * p<0.05. In panels A, C–E, data were analyzed using one-way ANOVA while data in panel B were analyzed using two-way ANOVA, and Tukey’s multiple comparison test was used for post hoc analysis.Figure 3. SNHG15 serves as a ceRNA for miR-18a to mediate THBS2 expression. (A, B) Sub-cellular localization of SNHG15 in N2a cells was determined by a RT-qPCR (A) and a FISH assay (B). (C, D) Binding relationships between miR-18a and SNHG15 (C), and between miR-18a and the 3′-UTR of CXCL13 mRNA (D) were predicted on Starbase (http://starbase.sysu.edu.cn/) and validated through dual-luciferase reporter gene assay. (E) Contents of total ERK/MEK and phosphorylated ERK/MEK in N2a cells was quantified by ELISA kits. F, phosphorylation of ERK1/2 and MEK1/2 was determined by Western blot analysis. Data were shown as mean±SD based on 3 independent experiments. * p<0.05. In panels A and F, data were analyzed using two-way ANOVA, while data in panels C–E were analyzed using one-way ANOVA, and Tukey’s multiple comparison test was used for post hoc analysis.Figure 4. Overexpression of miR-18a inhibits apoptosis and promotes viability of N2a cells. (A) MiR-18a expression before and after miR-18a mimic transfection was performed using RT-qPCR. (B) Viability of OGD-treated N2a cells was detected using EdU labeling assay. (C, D) Apoptosis of OGD-treated N2a cells was detected using Hoechst 33258 staining (C) and flow cytometry (D). Data are shown as mean±SD based on 3 independent experiments. * p<0.05. Data were analyzed using the unpaired t test.Figure 5. The ERK/MEK antagonist reverses the roles of SNHG15 siRNA and miR-18a mimic in N2a cells. An ERK/MEK antagonist, U0126, was introduced into N2a cells with silenced SNHG15 or overexpressed miR-18a. (A) Viability of N2a cells was detected using EdU labeling assay. (B, C) Apoptosis of OGD-treated N2a cells was detected using Hoechst 33258 staining (B) and flow cytometry (C), respectively. Data are shown as mean±SD based on 3 independent experiments. * p<0.05. Data were analyzed using one-way ANOVA. In Press
Clinical Research
Effect of Indirect Co-Culture With Gingival Mesenchymal Stem Cells on Cytokine Secretion in Primary Oral Sq...Med Sci Monit In Press; DOI: 10.12659/MSM.952439
Clinical Research
Comparison of Sleep Architecture in Individuals Aged 65 to 80 Years With and Without Mild Cognitive Impairm...Med Sci Monit In Press; DOI: 10.12659/MSM.952493
Clinical Research
Effects of Single-Bout Endurance Exercise Intensity on Peripheral Neurotrophic Factors in Patients With Isc...Med Sci Monit In Press; DOI: 10.12659/MSM.952089
Review article
Anisodus tanguticus in Cancer Research: A Review of Traditional Use, Phytochemistry, Extraction Methods, an...Med Sci Monit In Press; DOI: 10.12659/MSM.952999
Most Viewed Current Articles
17 Jan 2024 : Review article 14,176,514
Vaccination Guidelines for Pregnant Women: Addressing COVID-19 and the Omicron VariantDOI :10.12659/MSM.942799
Med Sci Monit 2024; 30:e942799
13 Nov 2021 : Clinical Research 3,760,677
Acceptance of COVID-19 Vaccination and Its Associated Factors Among Cancer Patients Attending the Oncology ...DOI :10.12659/MSM.932788
Med Sci Monit 2021; 27:e932788
14 Dec 2022 : Clinical Research 2,466,264
Prevalence and Variability of Allergen-Specific Immunoglobulin E in Patients with Elevated Tryptase LevelsDOI :10.12659/MSM.937990
Med Sci Monit 2022; 28:e937990
16 May 2023 : Clinical Research 708,906
Electrophysiological Testing for an Auditory Processing Disorder and Reading Performance in 54 School Stude...DOI :10.12659/MSM.940387
Med Sci Monit 2023; 29:e940387