20 August 2021: Animal Study
Knockout of the Cannabinoid Receptor 2 Gene Promotes Inflammation and Hepatic Stellate Cell Activation by Promoting A20/Nuclear Factor-κB (NF-κB) Expression in Mice with Carbon Tetrachloride-Induced Liver Fibrosis
Cuizhen Long123ABCDEF, Na Xie12ABCDEF, Yuanhui Shu12ABCF, Yafeng Wu14ABCF, Ping He12BC, Yan Zhou12BC, Yining Xiang5CD, Junying Gu12CD, Lei Yang12DF, Yuping Wang12ABCDEFG*DOI: 10.12659/MSM.931236
Med Sci Monit 2021; 27:e931236






Abstract
BACKGROUND: This study aimed to investigate the effect of deleting the cannabinoid receptor 2 (CB2) gene on the development of hepatic fibrosis induced by carbon tetrachloride (CCl₄) in mice via regulating inflammation.
MATERIAL AND METHODS: The DNA was extracted from the tails of mice to identify whether the cannabinoid receptor 2 gene was successfully knocked out. A liver fibrosis model was established by an intraperitoneal injection of CCl₄ into mice. Hepatic damage and hepatic fibrosis were evaluated by detecting serum alanine aminotransferase (ALT), aspartate aminotransferase (AST), and staining paraffin sections of liver tissue with hematoxylin-eosin (HE). The secretion and distribution of collagen in liver tissue were observed by Masson staining. Western blot analysis was performed to detect the expression of a-smooth muscle actin (α-SMA), transforming growth factor-β1 (TGF-β1), tumor necrosis factor alpha-induced protein 3 (A20), phosphorylated nuclear factor-kB p65 (p-NF-κB p65), tumor necrosis factor alpha (TNF-α), and interleukin-6 (IL-6) in liver tissue. Reverse transcription-polymerase chain reaction (RT-PCR) was used to detect the expression of IL-6 and TNF-α mRNA in liver tissue.
RESULTS: Compared with the control mice, the mice with CB2 knockout that were exposed to CCl₄ exhibited increased liver damage, liver fibrosis, and upregulated α-SMA, TGF-β1, A20, and p-NF-κB p65 protein levels. IL-6 and TNF-α protein levels and mRNA levels were upregulated.
CONCLUSIONS: The deletion of the CB2 gene promoted the activation of hepatic stellate cells in mice with liver fibrosis and aggravated liver fibrosis by up-regulating the protein expression of A20 and p-NF-κB p65 and inducing inflammatory response, potentially providing new insight into the treatment of liver fibrosis.
Keywords: Cannabinoid Receptor 2 Gene, A20, liver fibrosis, NF-κB, Carbon Tetrachloride, Collagen, Disease Susceptibility, Gene Expression Regulation, hepatic stellate cells, Immunohistochemistry, Liver, Liver Cirrhosis, Receptor, Cannabinoid, CB2, Tumor Necrosis Factor alpha-Induced Protein 3
Background
Liver fibrosis is a pathological process in which tissue damage and repair occur simultaneously after chronic liver injury. The primary mechanism of liver fibrosis is scar formation. Oxidative stress, tissue hypoxia, inflammation, and immune response cause massive hepatocyte necrosis. If these factors stimulate the liver for a long time, it will cause the rate of hepatocyte necrosis to exceed the rate of hepatocyte regeneration. In the process, hepatic stellate cells (HSCs) are activated and produce a large amount of extracellular matrix (ECM) that is deposited in liver tissue instead of hepatocytes [1]. The transformation of activated hepatic stellate cells into hepatic myofibroblasts plays a key role in the progression of hepatic fibrosis. HSCs are silent in normal liver tissue but are activated by hepatic injury, and transform into myofibroblast-like cells during the fibrotic process. Increased expression of α-SMA is a marker of activated hepatic stellate cells [2]. Activated HSCs secrete transforming growth factor (TGF-β1), which induces collagen production that leads ECM accumulation [3]. TGF-β1 is present in normal liver tissue, but increased expression occurs at all stages of progressive liver disease, including fibrosis [4]. Production of TGF-β1 contributes to activation of HSCs and excessive accumulation of ECM, thereby promoting liver fibrosis [5]. The secretion of various inflammatory factors, such as interleukin-6 (IL-6) and tumor necrosis factor alpha (TNF-α), further promote the activation of HSCs, which results in deterioration of liver fibrosis [1,6]. Focusing on the link between the inflammatory response and the activation of HSCs will help us further explore the mechanisms by which liver fibrosis occurs.
A20, also known as tumor necrosis factor alpha-induced protein 3 (TNFAIP3), is a cytoplasmic zinc finger protein that encodes an ubiquitin-editing enzyme. A20 plays a key role in negative regulation of the inflammatory response [7]. The anti-inflammatory function of A20 was further supported in several other inflammatory diseases, such as bronchial asthma, rheumatoid arthritis, and myocarditis [8–10]. Wang Xiaohan et al showed that A20 can alleviate liver fibrosis in non-alcoholic fatty liver disease (NAFLD) by inhibiting activation of hepatic stellate cells and production of inflammatory mediators in liver tissue [11].
As research has progressed, an increasing number of studies have shown that the endogenous cannabinoid system (ECS) plays an important role in liver fibrosis [12]. The ECS mainly consists of cannabinoid receptors, endogenous ligands, and enzymes related to ligand synthesis and degradation [13]. Among these components, the cannabinoid receptor 2 (CB2) is one of the most important components of the ECS. Many studies have shown that CB2 agonists can reduce the severity of portal hypertension, portal choroid plexus, and mesenteric angiogenesis in cirrhotic rats with intrahepatic angiogenesis and fibrosis [14]. Moreover, the activation of endogenous CB2 in hepatic fibroblasts can reduce experimental liver fibrosis [15], and CB2 can reduce the extent of liver damage in acute liver injury caused by ischemia-reperfusion or concanavalin [16,17]. Our previous study also showed that the CB2 agonist AM1241 can inhibit the proliferation of the HSC-T6 cell line cultured in vitro by protecting against oxidative stress and reducing the production of ECM [18]; in vivo, our previous experiments showed that the CB2 agonist AM1241 can ameliorate carbon tetrachloride (CCl4)-induced liver fibrosis in mice by mediating the expression of platelet-derived growth factor (PDGF), thereby inhibiting the activation of HSCs to prevent the progression of liver fibrosis [19]. Many of these experimental results indicated that the CB2 receptor has anti-fibrosis effects. There was also a study showing that administration of CCl4 to CB2−/− mice accelerated liver injury, as shown by increased alanine/aspartate aminotransferase levels and hepatocyte apoptosis [20].
However, the specific mechanism of cannabinoid receptor 2 affecting liver fibrosis remains to be elucidated. Although there have been many studies on cannabinoid receptor 2 or A20 in the past, whether the effect of cannabinoid receptor 2 on liver fibrosis is related to the A20/NF-κB p65 pathway has not been reported. Many studies have concluded that cannabinoid receptor 2 or A20 may be a potential target for the treatment of liver fibrosis. We study 2 targets at the same time; if their joint effect can further improve liver fibrosis, it will undoubtedly provide a new strategy for the prevention and treatment of liver fibrosis. Therefore, here, we investigated the effect of deleting the cannabinoid receptor 2 (CB2) gene on the development of hepatic fibrosis induced by carbon tetrachloride (CCl4) in mice via regulating A20/NF-κB expression.
Material and Methods
ANIMALS:
Clean-grade wild-type (WT) C57BL/6J mice were purchased from the Animal Experimental Center of Guizhou Medical University, and CB2−/− C57BL/6J mice were purchased from Jackson Laboratory, USA. All animal experiments were approved by the Guizhou Medical University Animal Care and Use Committee.
IDENTIFICATION OF CANNABINOID RECEPTOR 2 GENE KNOCKOUT:
Total DNA was extracted from the tails of mice using the DNA extraction reagent (Beijing Tiangen, China) according to the manufacturer’s protocol and was subsequently quantified using the NanoDrop™ 2000 device (Thermo Fisher Scientific, USA), according to the manufacturer’s protocol. The primers of cannabinoid receptor 2 gene were designed by Jackson Laboratory, USA and were synthesized by Shanghai Biological Engineering of China. The primer sequences are listed in Table 1. PCR was performed using MightAMP™ DNA Polymerase Ver.3 (TaKaRa, Japan) according to the manufacturer’s protocol. The thermal cycling conditions for PCR were: 98°C for 2 min, 98°C for 20 s +58°C for 15 s +72°C for 35 s (35 cycles), then held at 4°C. PCR amplification products were analyzed by 1.5% agarose gel electrophoresis, then were observed in a gel imager (Bio-Rad, USA).
ESTABLISHMENT OF LIVER FIBROSIS MODEL:
Twelve 8-week-old WT C57BL/6J male mice were randomly divided into 2 groups, the WT control group and the WT model group, with 6 mice in each group. Twelve 8-week-old CB2−/− C57BL/6J male mice were randomly divided into 2 groups, the CB2−/− control group and the CB2−/− model group, with 6 mice in each group. The mice in the WT model group and the CB2−/− model group were intraperitoneally injected with CCl4 (Aladdin, China) at 5 ml/kg (diluted with edible olive oil before injecting). The dose in the first week was 10%, and the dose in the second week was adjusted to 30%. CCl4 was administered 3 times per week for 16 weeks. The 2 control groups were administered the same solvent in the same manner. After 16 weeks, the mice were anaesthetized, the eyeballs of mice were removed to collect blood, and the liver tissues were dissected. Some liver tissue samples were fixed in 4% formaldehyde, and the remaining samples were stored at −80°C until use. Serum ALT and AST levels were measured on an automatic biochemical analyzer (Guizhou Jinyu Medical Testing Centre Limited Corporation, Guiyang, China).
HISTOPATHOLOGICAL ANALYSIS:
After the liver tissue was fixed in 4% formaldehyde for 24 h, paraffin-embedded liver tissue sections were obtained through dehydration, transparentizing, and paraffin embedding. HE and Masson staining were carried out on paraffin-embedded liver tissue sections. The pathological changes in the liver tissue were observed under a light microscope.
WESTERN BLOT ANALYSIS:
Total protein from the liver tissue was extracted using a whole protein extraction kit (Suo Laibao, China). Total protein content was determined using a BCA Assay kit (Suo Laibao, China). Protein (20 μg) was loaded and separated by SDS-PAGE electrophoresis, which was transferred to a PVDF membrane. Membranes were subsequently blocked using 5% skimmed milk with TBST at room temperature for 2 h and incubated with the following primary antibodies at 4°C overnight: α-SMA (Abcam, USA); TGF-β1 (Abcam, USA); A20 (Cell Signaling Technology, USA); p-NF-κB p65 (Cell Signaling Technology, USA); NF-κB p65 (Cell Signaling Technology, USA); TNF-α (Abcam, USA); IL-6 (Abcam, USA); and GAPDH (Abcam, USA). The membrane was then incubated with goat anti-rabbit IgG H&L secondary antibody at room temperature for 1.5 h. The signal on the membrane was detected using an enhanced chemiluminescence reagent (Millipore, USA) and a chemiluminescence instrument (Clinx, China) was used for quantification. Image J software was used to analyze the grey value of all proteins.
REAL-TIME REVERSE TRANSCRIPTION-POLYMERASE CHAIN REACTION:
Total RNA was extracted from the liver tissue using the RNA extraction reagent (Axygen, USA) according to manufacturer’s protocol and subsequently quantified using a NanoDrop™ 2000 machine (Thermo Fisher Scientific, USA), according to the manufacturer’s protocol. The total RNA was reverse-transcribed to synthesize cDNA using the Takara PrimeScript RT Master Mix kit (TaKaRa Bio, Japan) according to manufacturer’s protocol. The primers of GAPDH, TNF-α, and IL-6 were designed by Shanghai Biological Engineering of China. The primer sequences are listed in Table 2. QPCR was performed using Power UP™ SYBR Green Master Mix (Life, USA) according to the manufacturer’s protocol. The thermal cycling conditions for QPCR were: 50°C for 2 min, 95°C for 2 min, 95°C for 15 s +60°C for 1 min (40 cycles), 95°C for 15 s, 60°C for 1 min, 95°C for 15 s. The mRNA expression levels of TNF-α and IL-6 were normalized to that of GAPDH and quantified based on the comparative cycle threshold Ct method (2−ΔΔCt).
STATISTICAL ANALYSIS:
All the values are represented as the mean±SD. Statistical analysis of the data was performed by analysis of variance (ANOVA) for multiple comparisons. The analysis was conducted using GraphPad Prism 5 software.
Results
SUCCESSFUL CONSTRUCTION OF CANNABINOID RECEPTOR 2 KNOCKOUT MICE:
The DNA product size of CB2−/− mice is about 550 bp (Figure 1), and the size of WT mice DNA product is about 385 bp (Figure 1). The negative control group (without any primers) had no expression of any products, and both products of the hybrid mice were expressed (Figure 1).
DELETION OF CANNABINOID RECEPTOR 2 AGGRAVATED LIVER DAMAGE:
Compared with the control group, the 2 model groups had significantly increased serum ALT and AST (P<0.05; Table 3), suggesting that the 2 model groups had serious liver damage. Compared with the WT model group, the CB2−/− model group had significantly increased serum ALT and AST (P<0.05; Table 3), indicating that the liver damage of mice was aggravated after knocking out the CB2 receptor gene.
DELETION OF CANNABINOID RECEPTOR 2 AGGRAVATED LIVER DAMAGE AND INFLAMMATORY CELL INFILTRATION:
There was no significant difference in the pathological changes between the WT control group and the CB2−/− control group; the structure of the hepatic lobule was intact and clearly visible. The central vein was located between the hepatic lobules, and the hepatocytes (black arrow) were arranged radially along the central vein. The cell membrane of hepatocytes was intact, the nucleus was located in the center, and there were no inflammatory cells (red arrow) infiltrating between the hepatic lobules (Figure 2). In the WT model group, the hepatic lobular structure disappeared, the pseudolobule was formed, many hepatocytes were necrotic, and there were many inflammatory cells infiltrating into liver tissue (Figure 2). Compared with the WT model group, the CB2−/− model group showed a significant increase in hepatocyte necrosis, and many inflammatory cells around the central vein infiltrated into a sheet (Figure 2).
DELETION OF CANNABINOID RECEPTOR 2 PROMOTED COLLAGEN SECRETION:
There was no significant difference in the pathological changes between the WT control group and the CB2−/− control group: liver cells (black arrow) were arranged neatly along the central vein, and there was almost no blue collagen fiber (blue arrow) (Figure 3). Compared with the WT control group, many blue collagen fibers were distributed around the central vein in the WT model group (Figure 3). Compared with the WT model group, the CB2−/− model group had more necrotic hepatocytes, and the blue collagen fibers are more widely distributed (Figure 3).
DELETION OF CANNABINOID RECEPTOR 2 PROMOTED THE ACTIVATION OF HSCS:
There was no statistically significant difference in the protein expression of α-SMA and TGF-β1 between the WT control group and the CB2−/− control group. Compared with the corresponding control group, the 2 model groups had significantly increased expression of α-SMA and TGF-β1 (P<0.05, Figure 4A, 4B). The expressions of α-SMA and TGF-β1 in the CB2−/− model group were increased compared with the WT model group (P<0.05, Figure 4A, 4B).
DELETION OF CANNABINOID RECEPTOR 2 PROMOTED THE EXPRESSION OF A20 AND P-NF-κB P65:
The protein expression of A20 and p-NF-κB p65 was not significantly different between the WT control group and the CB2−/− control group (Figure 5A, 5B). Compared with the corresponding control group, the 2 model groups had significantly increased expression of A20 and p-NF-κB p65 (P<0.05, Figure 5A, 5B). In the CB2−/− model group, the expressions of A20 and p-NF-κB p65 were significantly upregulated when compared with the WT model group (P<0.05, Figure 5A, 5B).
DELETION OF CANNABINOID RECEPTOR 2 AGGRAVATED THE INFLAMMATORY RESPONSE BY PROMOTING IL-6 AND TNF-α MRNA AND PROTEIN LEVELS:
The mRNA expression of IL-6 and TNF-α were not significantly different between the WT control group and the CB2−/− control group (Figure 6A, 6B). IL-6 and TNF-α mRNA levels in the 2 model groups were significantly higher compared with the corresponding control group (P<0.05; Figure 6A, 6B); IL-6 and TNF-α mRNA levels were significantly increased in the CB2−/−model group compared with the WT model group (P<0.05; Figure 6A, 6B). The results of the proteins of IL-6 and TNF-α were consistent with those of mRNA (Figure 6C, 6D)
Discussion
Chronic liver disease, including viral, chemical, drug, immune, alcoholic, and cholestatic liver disease, can lead to liver fibrosis [21]. CCl4 is the most widely used chemical liver poison to induce liver fibrosis and cirrhosis in experimental animals. The long-term administration of CCl4 intraperitoneal injection into mice can permanently destroy hepatocytes through oxidative stress and inflammatory response, then activate quiescent HSCs, which subsequently secrete a large amount of ECM that gets deposited in the liver [22], thereby causing liver fibrosis in mice.
A large number of studies have shown that activate CB2 has anti-fibrosis effects. Mallat et al found that CB2 is upregulated in liver tissue of cirrhosis and that the activation of CB2 can delay the progression of fibrosis [23]. In mice exposed to CCl4, CB2 antagonist is manifested by increased fibrosis, while CB2 prevents fibrosis [24]. Our previous results have demonstrated that cannabinoid receptor agonists can inhibit the activation of HSCs in vivo and in vitro, delaying the development of liver fibrosis [18,19], which is consistent with the above findings. In addition, in animal models, the loss of CB2 results in collagen deposition, steatosis, and increased inflammation [25]. Eixeira-Clerc et al also demonstrated that administration of CCl4 to mice with knocked-out CB2 receptors can accelerate liver damage [15]. However, there have been no further studies on how the deletion of CB2 receptors aggravates liver fibrosis. Our other previous experiments showed that the deletion of the cannabinoid receptor 2 gene can aggravate concanavalin-induced acute liver injury by promoting the proliferation and activation of mouse macrophages [26]. Therefore, it is necessary to verify that knockout of the CB2 gene can aggravate liver fibrosis, and then to study the possible mechanism by which CB2 receptor deficiency aggravates liver fibrosis. The following results were obtained: the liver fibrosis model was established successfully; the degree of liver damage was aggravated and the activation of HSCs was increased in the liver fibrosis model groups compared with the control groups; the protein levels of A20, p-NF-κB p65, IL-6, and TNF-α expression were increased after the mouse CB2 gene was knocked out; the mRNA of IL-6 and TNF-α levels also were upregulated in the CB2−/− model group. These results suggest that the absence of CB2 aggravates hepatic stellate cell activation and inflammatory response through promoting A20/NF-κB p65 expression, ultimately aggravating liver fibrosis.
Detecting liver-associated biomarkers in the serum and staining liver tissue sections with HE are the 2 most direct methods for determining liver function damage. The pro-hepatic toxin CCl4 is directly injected into the liver by intraperitoneal injection, directly destroying liver cells, which release of a large amount of ALT and AST that can be detected in the serum. According to the serum ALT and AST detection and HE staining results, the serum ALT and AST levels in the 2 model groups were significantly higher than those in the control group. HE staining revealed significant necrosis of hepatocytes, loss of hepatic lobule structure, and infiltration of a large number of inflammatory cells. These results indicated that liver injury was serious and that the liver fibrosis model was successfully replicated. Compared with the WT model group, the CB2−/− model group had significantly increased ALT and AST levels; moreover, more extensive hepatocyte necrosis and more severe inflammatory cell infiltration were observed in the CB2−/− model group than in the WT model group, indicating that deletion of the CB2 gene aggravates liver fibrosis.
CB2 is mainly expressed in immune cells (B cells, natural killer cells, monocytes, macrophages, and neutrophils) and is also expressed in liver non-parenchymal cells such as HSCs and hepatic macrophages [15,27]. Under the stimulation of various liver injuries, pathogenic factors or platelet-derived growth factor (PDGF), transforming growth factor-β1 (TGF-β1), tumor necrosis factor-α (TNF-α), and interleukin-6 (IL-6), the activation of quiescent HSCs is the core mechanism of hepatic fibrosis, which is characterized by expression α-SMA and secreting a large amount of ECM mainly composed of type I and type III collagen [1,6,28]. TGF-β1 is the most important cytokine that is currently known to cause liver fibrosis [4]. Under normal conditions, the expression of TGF-β1 in liver is minimal, and its expression is significantly increased in liver injury. TGF-β1 can promote the expression of tissue inhibitor of metalloproteinase (TIMP) and collagen fibers, thereby activating HSCs [29]. Compared with the WT model group, the CB2−/− model group had increased expression of α-SMA and TGF-β1 proteins in liver tissue, as well as significantly increased secretion of blue collagen fibers, as evidenced by Masson staining. The above results fully illustrate the effect of the CB2 gene on HSCs: the deletion of the CB2 gene can promote activation of HSCs in the liver tissue of mice with liver fibrosis, thereby accelerating the progression of liver fibrosis. This also proves that CB2 receptor can inhibit the activation of hepatic stellate cells in liver tissue and plays a role in anti-fibrosis.
NF-κB is a nuclear transcription factor, and inactivated NF-κB is present in the cytoplasm. Upon activation, NF-κB is phosphorylated and transferred to the nucleus, where it is involved in cell proliferation, inflammation, and immune regulation [30]. When NF-κB is activated, it can initiate the inflammatory reaction by inducing the expression of tumor necrosis factor (TNF), interleukin-1 (IL-1), and cyclooxygenase 2, and the increased expression of these inflammatory factors promotes the activation of NF-κB, forming a positive feedback loop that activates NF-κB [31]. Zhao Zonghao et al showed that NF-κB mainly regulates the release of inflammatory mediators in Kupffer cells (KCs) during the early stage of liver fibrosis and has an important pro-inflammatory effect. In the late stage of liver fibrosis, NF-κB mainly regulates HSC activation to promote fibrosis formation [32]. Our results are consistent with the above studies: the protein of p-NF-κB p65 level in the liver fibrosis model group was higher than that in the control group, and the levels of the inflammatory cytokines IL-6 and TNF-α were also increased in the live fibrosis model group compared with the control group, suggesting that NF-κB can promote the inflammatory response. The inflammatory response of the CB2−/− model group was more prominent than that of the WT model group, indicating that the CB2 gene regulates the inflammatory response during liver fibrosis through NF-κB. In the process of osteoarthritis, atherosclerosis and ischemia-reperfusion injury, the activation of CB2 plays an important anti-inflammatory role, as CB2 is involved in inhibiting the activation, proliferation and migration of immune cells, and in inhibiting the expression of inflammatory factors (IL-1β, IL-6, TNF-α) [33].
In addition, A20 (TNFAIP3) is considered to be an important anti-inflammatory signaling molecule and a key negative regulator of the NF-κB signaling pathway [11]. Ubiquitin-editing protein A20 can reduce inflammation and protect hepatocytes from damage by inhibiting NF-κB activation [34]. Since A20 is an important regulator of inflammatory signaling, which is mainly involved the powerful termination of NF-κB activation [35,36] and is a crucial hepatoprotective factor in processes such as hepatic ischemia-reperfusion [37], liver regeneration and repair [38], acute toxic lethal hepatitis [39], and lethal radical hepatectomy [40], A20 level was elevated in patients with NASH [11], it is not surprising that A20 expression was upregulated in hepatic fibrosis specimens. Consistent with the above results, our study has also demonstrated that the expression of A20 is increased in hepatic fibrosis, and the expression of A20 was significantly higher in the CB2−/− model group when compared with the WT model group. These results support that A20 may be part of the physiological response to liver fibrogenesis. The loss of the CB2 gene aggravates liver inflammation, which leads to increased A20 reactivity, indicating that the CB2 gene plays an anti-fibrotic role, probably through the A20/NF-κB signaling pathway.
Conclusions
In summary, in this study, liver fibrosis model was successfully established in mice with CB2 knockout, in which liver tissue collagen fibers, α-SMA, TGF-β1, A20, p-NF-κB, TNF-α, and IL-6 protein levels, IL-6 and TNF-α mRNA levels were significantly changed. These results indicate that the inflammatory response is positively correlated with the activation of HSCs, thereby promoting liver fibrosis, and that the loss of the CB2 gene may be involved in the regulation of the inflammatory response through the A20/NF-κB signaling pathway in liver fibrosis. In the future, by combining in vivo and in vitro experiments, we will further explore the specific mechanism by which cannabinoid receptor 2 affects liver fibrosis.
Figures
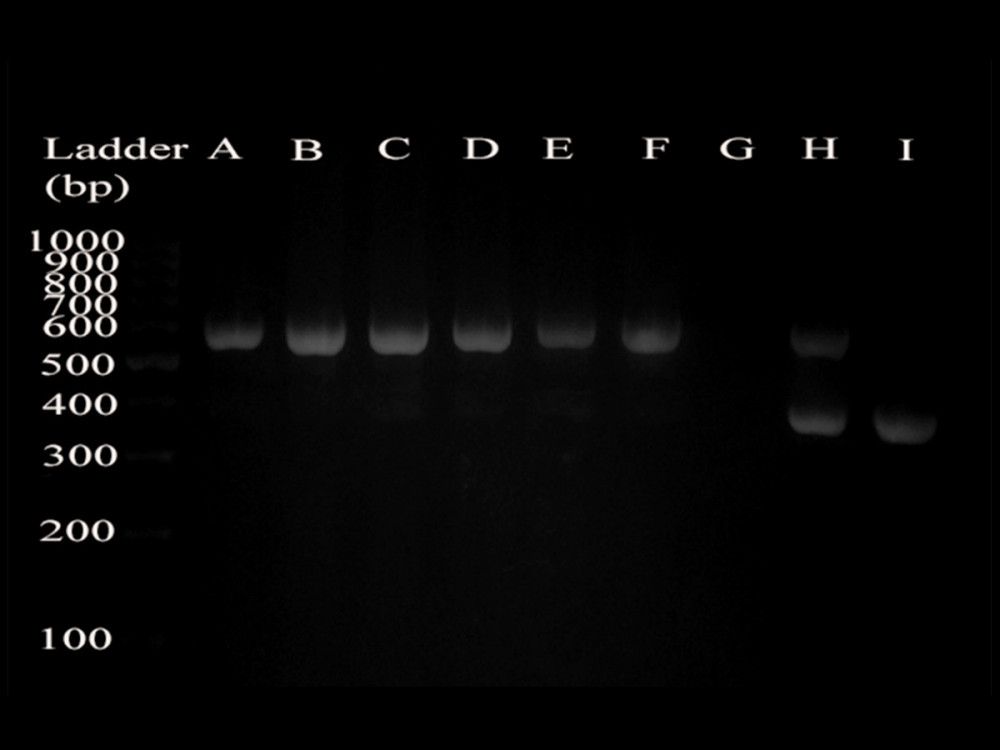 Figure 1. Identification of cannabinoid receptor 2 gene knockout. The extracted DNA from the tails of mice was subjected to PCR reaction and agarose gel electrophoresis, and the following results were obtained: the DNA band of CB2−/− mice (A, B, C, D, E, F) was about 550 bp, and the DNA band of WT mice (I) was about 385 bp. The negative control group (G) did not have any product bands, and the hybrid mice (H) had 2 bands (550 bp and 385 bp).
Figure 1. Identification of cannabinoid receptor 2 gene knockout. The extracted DNA from the tails of mice was subjected to PCR reaction and agarose gel electrophoresis, and the following results were obtained: the DNA band of CB2−/− mice (A, B, C, D, E, F) was about 550 bp, and the DNA band of WT mice (I) was about 385 bp. The negative control group (G) did not have any product bands, and the hybrid mice (H) had 2 bands (550 bp and 385 bp). 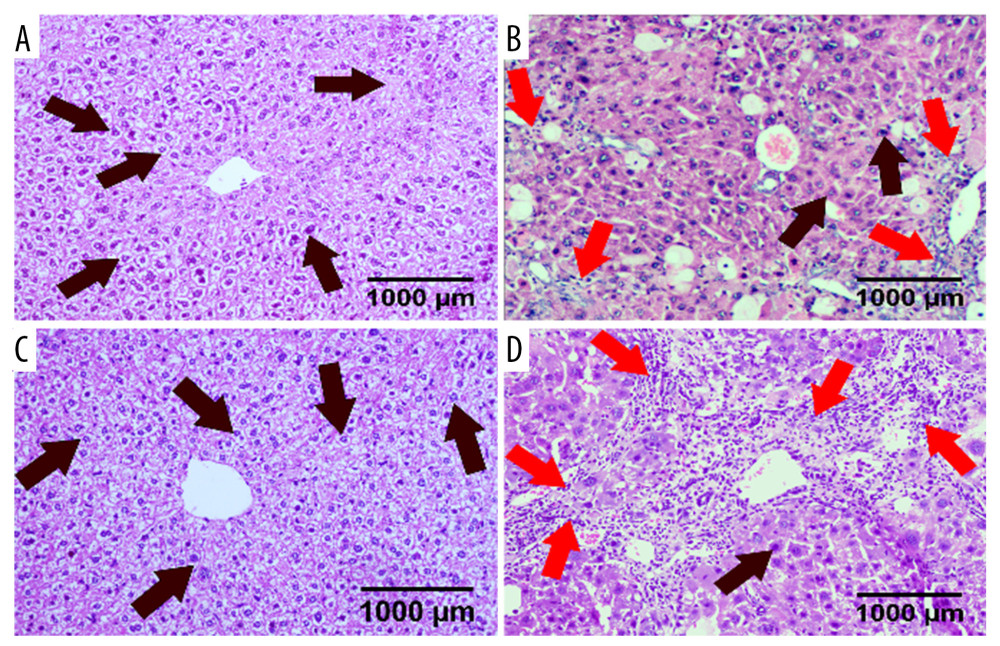 Figure 2. HE staining of liver sections in 4 groups of mice. Deletion of cannabinoid receptor 2 (CB2) aggravated liver damage and inflammatory cell infiltration. (A) WT control group; (B) WT model group; (C) CB2−/− control group; (D) CB2−/− model group. HE, hematoxylin-eosin.
Figure 2. HE staining of liver sections in 4 groups of mice. Deletion of cannabinoid receptor 2 (CB2) aggravated liver damage and inflammatory cell infiltration. (A) WT control group; (B) WT model group; (C) CB2−/− control group; (D) CB2−/− model group. HE, hematoxylin-eosin. 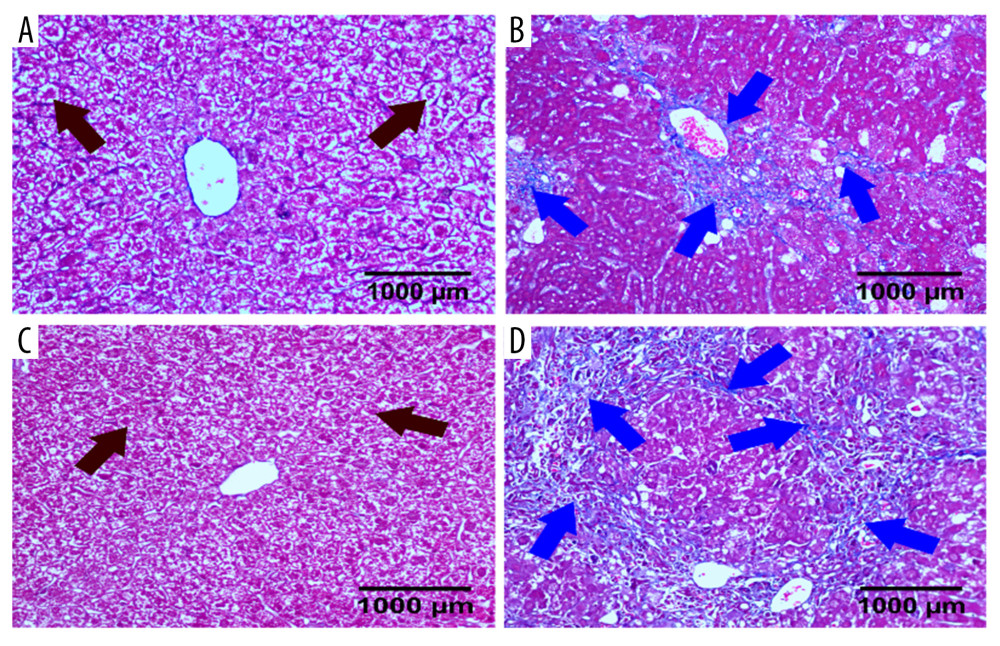 Figure 3. Masson staining of liver sections in 4 groups of mice. Deletion of cannabinoid receptor 2 (CB2) promoted collagen secretion. (A) WT control group; (B) WT model group; (C) CB2−/− control group; (D) CB2−/− model group.
Figure 3. Masson staining of liver sections in 4 groups of mice. Deletion of cannabinoid receptor 2 (CB2) promoted collagen secretion. (A) WT control group; (B) WT model group; (C) CB2−/− control group; (D) CB2−/− model group. 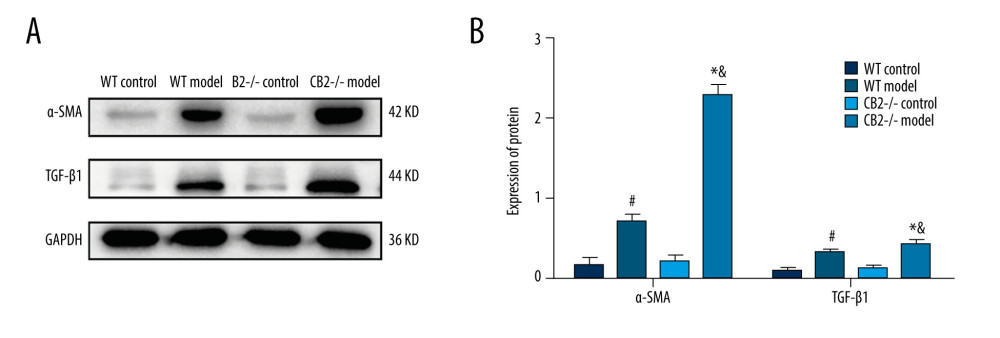 Figure 4. (A, B) Expression of α-SMA and TGF-β1 in liver tissue of 4 groups. Deletion of cannabinoid receptor 2 promoted the activation of HSCs. α-SMA, α-smooth muscle actin; TGF-β1, transforming growth factor-β1. # P<0.05 compared with the WT control group, * P<0.05 compared with the CB2−/− control group, and & P<0.05 compared with the WT model group.
Figure 4. (A, B) Expression of α-SMA and TGF-β1 in liver tissue of 4 groups. Deletion of cannabinoid receptor 2 promoted the activation of HSCs. α-SMA, α-smooth muscle actin; TGF-β1, transforming growth factor-β1. # P<0.05 compared with the WT control group, * P<0.05 compared with the CB2−/− control group, and & P<0.05 compared with the WT model group. 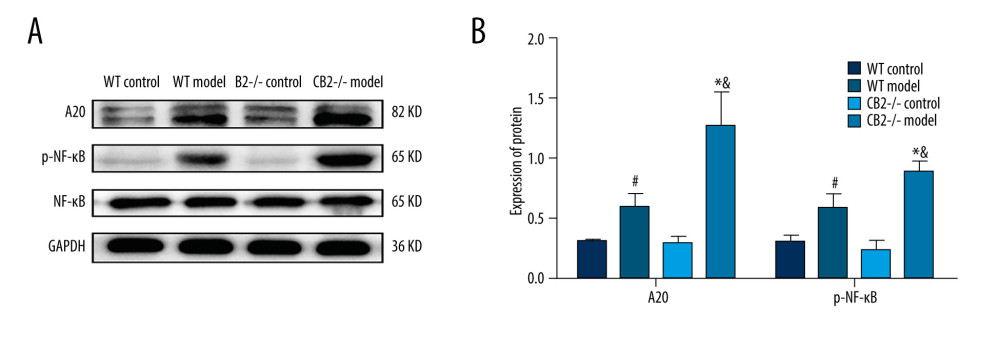 Figure 5. (A, B) Expression of A20 and p-NF-κB p65 in liver tissue of 4 groups. Deletion of cannabinoid receptor 2 promoted the expression of A20 and p-NF-κB p65 in liver tissue. A20, tumor necrosis factor alpha-induced protein 3; p-NF-κB p65, phosphorylated nuclear factor-κB p65. # P<0.05 compared with the WT control group, * P<0.05 compared with the CB2−/− control group, and & P<0.05 compared with the WT model group.
Figure 5. (A, B) Expression of A20 and p-NF-κB p65 in liver tissue of 4 groups. Deletion of cannabinoid receptor 2 promoted the expression of A20 and p-NF-κB p65 in liver tissue. A20, tumor necrosis factor alpha-induced protein 3; p-NF-κB p65, phosphorylated nuclear factor-κB p65. # P<0.05 compared with the WT control group, * P<0.05 compared with the CB2−/− control group, and & P<0.05 compared with the WT model group. 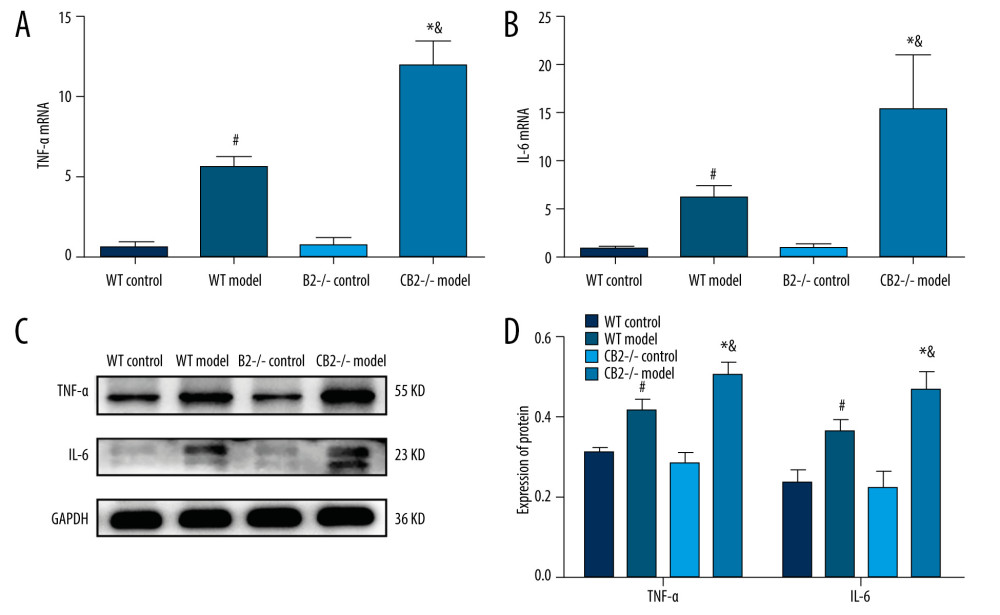 Figure 6. TNF-α and IL-6 mRNA and protein levels of liver tissue in 4 groups. The mRNA expression of TNF-α (A). The mRNA expression of IL-6 (B). The protein expression of TNF-α and IL-6 (C, D). TNF-α, tumor necrosis factor-α; IL-6, interleukin-6. # P<0.05 compared with the WT control group, * P<0.05 compared with the CB2−/− control group, and & P<0.05 compared with the WT model group.
Figure 6. TNF-α and IL-6 mRNA and protein levels of liver tissue in 4 groups. The mRNA expression of TNF-α (A). The mRNA expression of IL-6 (B). The protein expression of TNF-α and IL-6 (C, D). TNF-α, tumor necrosis factor-α; IL-6, interleukin-6. # P<0.05 compared with the WT control group, * P<0.05 compared with the CB2−/− control group, and & P<0.05 compared with the WT model group. Tables
Table 1. The primer sequences of CB2 gene used for PCR.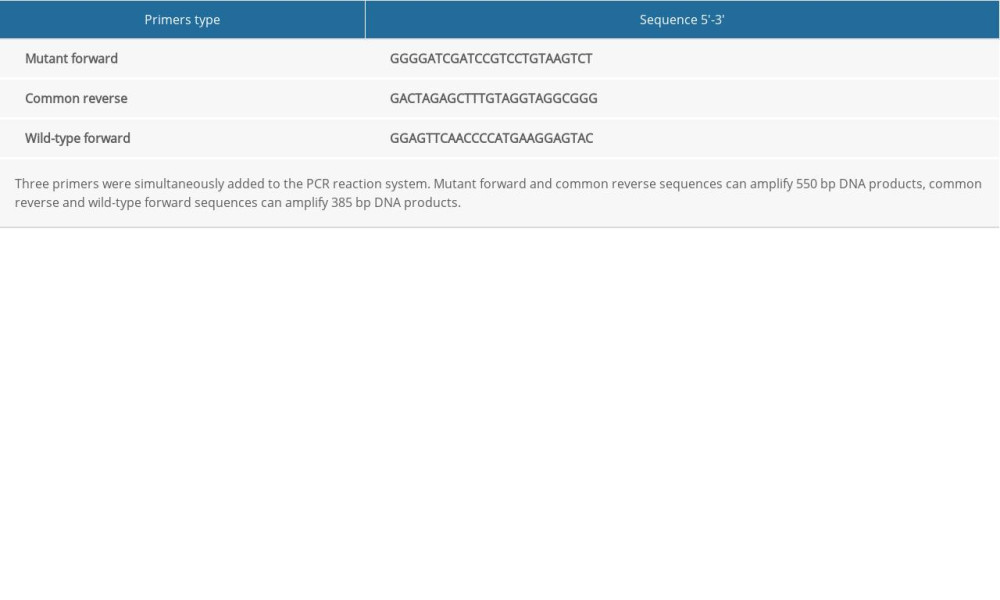 Table 2. Primer sequences used for reverse transcription-quantitative PCR.
Table 2. Primer sequences used for reverse transcription-quantitative PCR.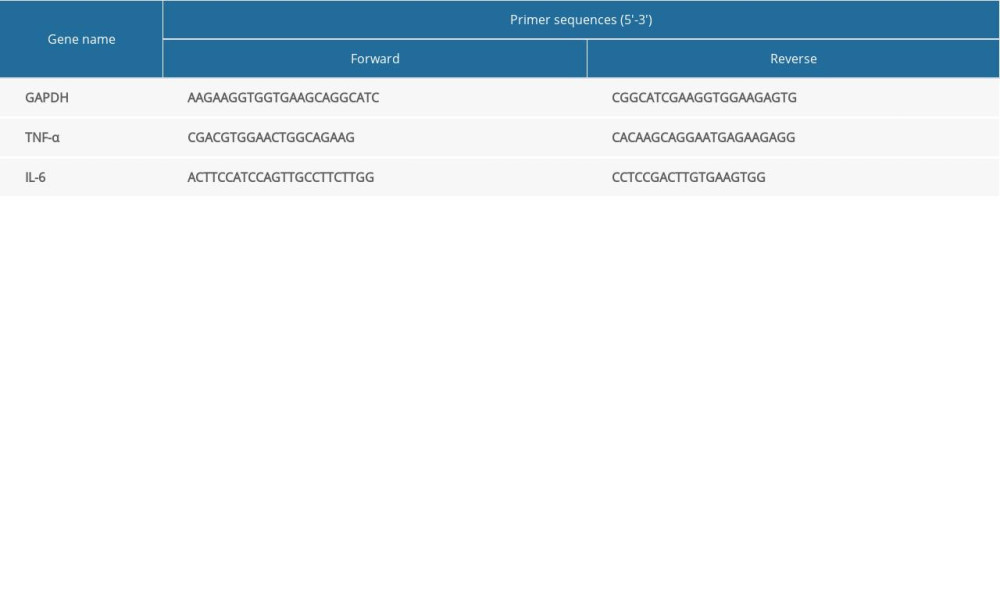 Table 3. Serum ALT and AST level values in 4 groups. Deletion of cannabinoid receptor 2 (CB2) aggravated liver damage. Serum ALT and AST level values are represented the mean±SD.
Table 3. Serum ALT and AST level values in 4 groups. Deletion of cannabinoid receptor 2 (CB2) aggravated liver damage. Serum ALT and AST level values are represented the mean±SD.
References
1. Ramos-Tovar E, Muriel P, Molecular mechanisms that link oxidative stress, inflammation, and fibrosis in the liver: Antioxidants (Basel), 2020; 9; 1279
2. Friedman SL, Hepatic stellate cells: Protean, multifunctional, and enigmatic cells of the liver: Physiol Rev, 2008; 88; 125-72
3. Wu L, Zhang Q, Mo W, Quercetin prevents hepatic fibrosis by inhibiting hepatic stellate cell activation and reducing autophagy via the TGF-β1/Smads and PI3K/Akt pathways: Sci Rep, 2017; 7; 9289
4. Dooley S, Dijke Pt, TGF-β in progression of liver disease: Cell Tissue Res, 2012; 347; 245-56
5. Xu F, Liu C, Zhou D, Zhang LTGF/SMAD pathways and its regulation in hepatic fibrosis: Journal of Histochemistry & Cytochemistry Official Journal of the Histochemistry Society, 2016; 64; 157 [in Chinese]
6. Higashi T, Friedman SL, Hoshida Y, Hepatic stellate cells as key target in liver fibrosis: Adv Drug Deliv Rev, 2017; 121; 27-42
7. Harhaj EW, Dixit VM, Regulation of NF-κB by deubiquitinases: Immunol Rev, 2012; 246; 107-24
8. Kang NI, Yoon HY, Lee YR, A20 attenuates allergic airway inflammation in mice: J Immunol, 2009; 183; 1488-95
9. Hah YS, Lee YR, Jun JS, A20 suppresses inflammatory responses and bone destruction in human fibroblast-like synoviocytes and in mice with collagen-induced arthritis: Arthritis Rheum, 2014; 62; 2313-21
10. Gui J, Yan Y, Ruizhen C, Wei X, Sidong X, A20 (TNFAIP3) alleviates CVB3-induced myocarditis via inhibiting NF-κB signaling: PLoS One, 2012; 7; e46515
11. Wang X, Ai L, Xu Q, A20 attenuates liver fibrosis in NAFLD and inhibits inflammation responses: Inflammation, 2017; 40; 840-48
12. Wang M, Meng N, Chang Y, Tang W, Endocannabinoids signaling molecular mechanisms of liver regulation and diseases: Front Biosci (Landmark Ed), 2016; 21; 1488
13. Pacher P, Kunos G, Modulating the endocannabinoid system in human health and disease – successes and failures: FEBS J, 2013; 280; 1918-43
14. Huang HC, Wang SS, Hsin IF, Cannabinoid receptor 2 agonist ameliorates mesenteric angiogenesis and portosystemic collaterals in cirrhotic rats: Hepatology, 2012; 56; 248-58
15. TeixeiraClerc F, Belot MP, Manin S, Beneficial paracrine effects of cannabinoid receptor 2 on liver injury and regeneration: Hepatology, 2010; 52; 1046-59
16. Bátkai S, Osei-Hyiaman D, Pan H, Cannabinoid-2 receptor mediates protection against hepatic ischemia/reperfusion injury: FASEB J, 2007; 21; 1788-800
17. Hegde VL, Hegde S, Cravatt BF, Attenuation of experimental autoimmune hepatitis by exogenous and endogenous cannabinoids: involvement of regulatory T cells: Mol Pharmacol, 2008; 74; 20-33
18. Shi Y, Wu YF, Long CZStudy of antioxidant effect of cannabinoid receptor type 2 agonist on rat hepatic stellate cell line: Zhonghua Gan Zang Bing Za Zhi, 2018; 26; 660-65 [in Chinees]
19. He P, Wu YF, Yang HYEffect of cannabinoid receptor-2 agonist AM1241 on platelet-derived growth factor expression in the liver tissue of mice with hepatic fibrosis: Zhonghua Gan Zang Bing Za Zhi, 2017; 25; 841-46 [in Chinees]
20. Julien B, Grenard P, Teixeira-Clerc F, Antifibrogenic role of the cannabinoid receptor CB2 in the liver: Gastroenterology, 2005; 128; 742-55
21. Altamirano-Barrera A, Barranco-Fragoso B, Méndez-Sánchez N, Management strategies for liver fibrosis: Ann Hepatol, 2017; 16; 48-56
22. Dong S, Chen QL, Song YN, Mechanisms of CCl4-induced liver fibrosis with combined transcriptomic and proteomic analysis: J Toxicol Sci, 2016; 41; 561-72
23. Mallat A, Teixeira-Clerc F, Deveaux V, The endocannabinoid system as a key mediator during liver diseases: New insights and therapeutic openings: Br J Pharmacol, 2011; 163; 1432-40
24. Baldassarre M, Giannone FA, Napoli L, The endocannabinoid system in advanced liver cirrhosis: Pathophysiological implication and future perspectives: Liver Int, 2013; 33; 1298-308
25. Patsenker E, Stickel F, Cannabinoids in liver diseases: Clin Liver Dis (Hoboken), 2016; 7; 21-25
26. Yafeng W, Cuizhen L, Yuanhui SCannabinoid receptor 2 deletion promotes proliferation and activation of hepatic macrophages in mice with acute liver injury induced by concanavalin A: Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi, 2019; 35; 13-18 [in Chinese]
27. Leleu-Chavain N, Desreumaux P, Chavatte P, Millet R: Curr Mol Pharmacol, 2013; 6; 183-203
28. Yin C, Evason KJ, Asahina K, Hepatic stellate cells in liver development, regeneration, and cancer: J Clin Invest, 2013; 123(5); 1902-10
29. Liu B, Zhang X, Zhang FC, Aberrant TGF-β1 signaling contributes to the development of primary biliary cirrhosis in murine model: World J Gastroenterol, 2013; 19; 5828-36
30. Lanucara F, Lam C, Mann J, Dynamic phosphorylation of RelA on Ser42 and Ser45 in response to TNFα stimulation regulates DNA binding and transcription: Open Biol, 2016; 6; 160055
31. Wang R, Zhang H, Wang Y, Inhibitory effects of quercetin on the progression of liver fibrosis through the regulation of NF-κB/IκBα, p38 MAPK, and Bcl-2/Bax signaling: Int Immunopharmacol, 2017; 47; 126-33
32. Zonghao ZH, Jianming X, Qiao MA contimuous observation of role of NF-κB in rats liver fibrosis: Chinese Journal of Clinical Health Care, 2010; 13(5); 496-98 [in Chinese]
33. Zhang J, Chen L, Su T, Electroacupuncture increases CB2 receptor expression on keratinocytes and infiltrating inflammatory cells in inflamed skin tissues of rats: J Pain, 2010; 11; 1250-58
34. Catrysse L, Farhang Ghahremani M, Vereecke L, A20 prevents chronic liver inflammation and cancer by protecting hepatocytes from death: Cell Death Dis, 2016; 7; e2250
35. Shembade N, Ma A, Harhaj EW, Inhibition of NF-κB signaling by A20 through disruption of ubiquitin enzyme complexes: Science, 2010; 327; 1135-39
36. Wertz IE, O’Rourke KM, Zhou H, De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling: Nature, 2004; 430; 694-99
37. Sun J, Sun L, Zhang N, A20 is up-regulated in primary mouse hepatocytes subjected to hypoxia and reperfusion: Cell Biochem Funct, 2012; 30(8); 683-86
38. Silva CG, Studer P, Skroch M, A20 promotes liver regeneration by decreasing SOCS3 expression to enhance IL-6/STAT3 proliferative signals: Hepatology, 2013; 57; 2014-25
39. Arvelo MB, Cooper JT, Longo C, A20 protects mice from D-galactosamine/lipopolysaccharide acute toxic lethal hepatitis: Hepatology, 2002; 35(3); 535-43
40. Longo CR, Patel VI, Shrikhande GV, A20 protects mice from lethal radical hepatectomy by promoting hepatocyte proliferation via a p21waf1-dependent mechanism: Hepatology, 2005; 42; 155-64
Figures
Figure 1. Identification of cannabinoid receptor 2 gene knockout. The extracted DNA from the tails of mice was subjected to PCR reaction and agarose gel electrophoresis, and the following results were obtained: the DNA band of CB2−/− mice (A, B, C, D, E, F) was about 550 bp, and the DNA band of WT mice (I) was about 385 bp. The negative control group (G) did not have any product bands, and the hybrid mice (H) had 2 bands (550 bp and 385 bp).Figure 2. HE staining of liver sections in 4 groups of mice. Deletion of cannabinoid receptor 2 (CB2) aggravated liver damage and inflammatory cell infiltration. (A) WT control group; (B) WT model group; (C) CB2−/− control group; (D) CB2−/− model group. HE, hematoxylin-eosin.Figure 3. Masson staining of liver sections in 4 groups of mice. Deletion of cannabinoid receptor 2 (CB2) promoted collagen secretion. (A) WT control group; (B) WT model group; (C) CB2−/− control group; (D) CB2−/− model group.Figure 4. (A, B) Expression of α-SMA and TGF-β1 in liver tissue of 4 groups. Deletion of cannabinoid receptor 2 promoted the activation of HSCs. α-SMA, α-smooth muscle actin; TGF-β1, transforming growth factor-β1. # P<0.05 compared with the WT control group, * P<0.05 compared with the CB2−/− control group, and & P<0.05 compared with the WT model group.Figure 5. (A, B) Expression of A20 and p-NF-κB p65 in liver tissue of 4 groups. Deletion of cannabinoid receptor 2 promoted the expression of A20 and p-NF-κB p65 in liver tissue. A20, tumor necrosis factor alpha-induced protein 3; p-NF-κB p65, phosphorylated nuclear factor-κB p65. # P<0.05 compared with the WT control group, * P<0.05 compared with the CB2−/− control group, and & P<0.05 compared with the WT model group.Figure 6. TNF-α and IL-6 mRNA and protein levels of liver tissue in 4 groups. The mRNA expression of TNF-α (A). The mRNA expression of IL-6 (B). The protein expression of TNF-α and IL-6 (C, D). TNF-α, tumor necrosis factor-α; IL-6, interleukin-6. # P<0.05 compared with the WT control group, * P<0.05 compared with the CB2−/− control group, and & P<0.05 compared with the WT model group. Tables
Table 1. The primer sequences of CB2 gene used for PCR.Table 2. Primer sequences used for reverse transcription-quantitative PCR.Table 3. Serum ALT and AST level values in 4 groups. Deletion of cannabinoid receptor 2 (CB2) aggravated liver damage. Serum ALT and AST level values are represented the mean±SD.Table 1. The primer sequences of CB2 gene used for PCR.Table 2. Primer sequences used for reverse transcription-quantitative PCR.Table 3. Serum ALT and AST level values in 4 groups. Deletion of cannabinoid receptor 2 (CB2) aggravated liver damage. Serum ALT and AST level values are represented the mean±SD. In Press
Clinical Research
Body Weight and Insulin Resistance Indicators Among ChildrenMed Sci Monit In Press; DOI: 10.12659/MSM.951434
Clinical Research
Comparison of Radiographic Cervical Sagittal Alignment Parameters in Patients With Nonspecific Neck Pain, D...Med Sci Monit In Press; DOI: 10.12659/MSM.952950
Clinical Research
Combined Fibrinogen and Urinary α1-Microglobulin as Predictors of Respiratory Tract Infection in Children w...Med Sci Monit In Press; DOI: 10.12659/MSM.951066
Database Analysis
Evaluation of Salivary Total Oxidant Status (TOS) and Total Antioxidant Status (TAS) in Orthodontic Patient...Med Sci Monit In Press; DOI: 10.12659/MSM.952052
Most Viewed Current Articles
17 Jan 2024 : Review article 14,175,576
Vaccination Guidelines for Pregnant Women: Addressing COVID-19 and the Omicron VariantDOI :10.12659/MSM.942799
Med Sci Monit 2024; 30:e942799
13 Nov 2021 : Clinical Research 3,756,620
Acceptance of COVID-19 Vaccination and Its Associated Factors Among Cancer Patients Attending the Oncology ...DOI :10.12659/MSM.932788
Med Sci Monit 2021; 27:e932788
14 Dec 2022 : Clinical Research 2,465,966
Prevalence and Variability of Allergen-Specific Immunoglobulin E in Patients with Elevated Tryptase LevelsDOI :10.12659/MSM.937990
Med Sci Monit 2022; 28:e937990
16 May 2023 : Clinical Research 708,651
Electrophysiological Testing for an Auditory Processing Disorder and Reading Performance in 54 School Stude...DOI :10.12659/MSM.940387
Med Sci Monit 2023; 29:e940387