08 December 2021: Lab/In Vitro Research
Chemokine (C-C Motif) Ligand 2/Chemokine Receptor 2 (CCR2) Axis Blockade to Delay Chondrocyte Hypertrophy as a Therapeutic Strategy for Osteoarthritis
Zidong Wang1ACE, Bei Wang2CF, Jian Zhang1DF, Zhensong Wu3BD, Liankui Yu1BF, Zhongye Sun1ADG*DOI: 10.12659/MSM.930053
Med Sci Monit 2021; 27:e930053






Abstract
BACKGROUND: Chondrocytes play a vital role in the later stages of osteoarthritis (OA). The roles of chemokine (C-C motif) ligand 2 (CCL2) and its receptor, chemokine receptor 2 (CCR2), are as yet poorly elucidated in chondrocyte hypertrophy (CH). Here, we aimed to regulate the CCL2/CCR2 axis and explore its effect on progression of CH.
MATERIAL AND METHODS: Chondrocytes isolated from patients with OA were used in the present study. In vitro experiments were conducted to test hypertrophic gene and CCL2/CCR2 expression in chondrocyte degeneration caused by interleukin (IL)-17A or CCL2 protein stimulation. In addition, inhibition of CCL2 and CCR2 was used to assess the role of CCL2 and CCR2 blockade in CH. Relative gene expression was determined with real-time polymerase chain reaction, western blot, or immunofluorescence. Hypertrophic changes were assessed with cell area measurement. Moreover, the viability of chondrocytes was analyzed using an MTT assay and flow cytometry was used to assess cell apoptosis.
RESULTS: CCL2 and CCR2 were upregulated in IL-17A-treated chondrocytes. The exogenic CCL2 stimulation also promoted CH and increased the expression of Type 10 collagen, RUNX2, and IHH, which could be reversed via suppression of CCR2. Inhibition of CCL2 and CCR2 expression was sufficient to: 1) protect Type 2 collagen synthesis; 2) alleviate IL-17A-induced overexpression of Type 10 collagen, RUNX2, and IHH; and 3) improve chondrocyte proliferation and apoptosis.
CONCLUSIONS: Blockading the CCL2/CCR2 axis plays a role in delaying the development of CH.
Keywords: Chondrocytes, Hypertrophy, Osteoarthritis, Chemokine CCL2, Female, Humans, Male, Receptors, CCR2, Signal Transduction
Background
Osteoarthritis (OA) is characterized by loss of cartilage, changes in subchondral bone, synovitis, and other joint tissue destruction. It is a chronic, degenerative disease that mainly affects weight-bearing joints; the typical clinical manifestations are joint pain, swelling, and limited activity [1,2]. Treatment of advanced OA is still mainly based on joint replacement, and there are currently no effective preventive measures. Cartilage hypertrophy can occur in normal embryonic chondrogenesis. However, chondrocytes that are hypertrophic express some typical biological markers, which mediate the continuing progression of OA [3]. As OA becomes more advanced, chondrocytes undergo differentiation, hypertrophic dedifferentiation, terminal mineralization, and, finally, apoptosis, which is similar to the differentiation of chondrocytes that occurs during ossification of cartilage. Disorders of chondrocyte growth regulation and a lack of factors that inhibit differentiation from maturation to hypertrophy accelerate OA development [4].
Chondrocytes in OA can produce a variety of inflammatory factors, such as interleukin (IL)-1β and tumor necrosis factor-α, which further promote the production of other inflammatory factors, thereby changing the composition of collagen and accelerating the decomposition of cartilage extracellular matrix (ECM), which is one of the leading causes of chondrocyte hypertrophy (CH) [5]. Chemokine (C-C motif) ligand 2 (CCL2), also known as MCP1, is an essential proinflammatory factor of the CC subfamily of chemokines [6,7]. It has a specific chemotactic activation effect on monocytes, macrophages, and other inflammatory factors that are commonly found in the synovial fluid of patients with OA [8]. The impact of chemokine-induced inflammation has been demonstrated in in vitro stimulation of hypertrophic chondrocyte differentiation related to
Chondrocytes, the only type of cells in the cartilage, play a vital role in the pathology of OA because of the character of the dedifferentiation and loss of the chondrogenic phenotype. Collagen type X is currently the central specific marker molecule for CH. Type II collagen is mainly present in the ECM of cartilage, which is secreted by normal chondrocytes and is a vital molecule marker of the chondrogenic phenotype. During the process of hypertrophy, production of Type II collagen in chondrocytes is significantly reduced, and the main components of the ECM change to Type X collagen, with increased levels of metalloproteinase-13 and proteoglycan enzymes, which promote cartilage calcification [14]. The cell surface receptors for CCL2 are C-C chemokine receptor 4 (CCR4) and chemokine receptor 2 (CCR2); CCR4 is a receptor for various chemokine CC subfamily members, but CCR2 mainly recognizes CCL2 [15]. We hypothesized that the CCL2-CCR2 ligand-receptor axis might play a role in the development of CH. The aim of the present study was to clarify the potential role of the CCL2/CCR2 axis in OA by examining Type 10 collagen and other specific molecular markers of hypertrophy [16].
Material and Methods
REAGENTS:
The Dulbecco’s modified Eagle’s medium (DMEM), fetal bovine serum (FBS), and bovine serum albumin (BSA) were from Gibco (Dun Laoghaire, Dublin, Ireland). The human recombinant interleukin (IL)-17A, penicillin-streptomycin mixture, trypsin, Type XI collagenase, and MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide) assay kits were from Sigma (St. Louis, Missouri, United States). The human recombinant CCL2 protein (Cat: 10134-H08Y) was from Sino Biological (Beijing, China). The CCR2 antagonist (CAS, Cat: 445479-97-0) was from Santa Cruz (Santa Cruz, California, United States). The bindarit (Bin) (Cat: S3032) was from Selleck (Shanghai, China). The anti-Type 10 collagen, anti-CCR2, anti-CCL2, and anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) primary antibodies were from Abcam (Cambridge, United Kingdom). The Alexa Fluor488 and 568, TRIzol reagent, and Annexin V-FITC/PI kit were from Invitrogen (Carlsbad, California, United States). The universal polymerase chain reaction (PCR) Master Mix was from TOYOBO (Osaka, Japan). Unless specifically indicated, all of the other reagents were from Beyotime (Shanghai, China).
CARTILAGE TISSUE COLLECTION:
Human cartilage was collected from patients with OA who were undergoing total knee arthroplasty at Liaocheng People’s Hospital. Informed consent was obtained from the patients before surgery. The study protocol was approved by the hospital’s Ethics Committee. A total of 23 patients donated their knee joints. Before surgery, cartilage with mild degeneration was obtained from 6 of them: 3 men and 3 women; average age 49 years, range 38 to 73 years. The inclusion criterion was absence of primary diseases other than OA, such as diabetes, hypertension, and cancer. The tissues were conserved in sterile growth medium immediately after they were cut from the patients and transferred to the laboratory for chondrocyte isolation.
CHONDROCYTE ISOLATION AND CULTURE:
We divided the superficial part of the cartilage to isolate the chondrocytes (n=6). The samples were washed with sterile phosphate-buffered saline (PBS) and cut into pieces as small as possible. The diced cartilage was then incubated with 0.15% trypsin and 0.2% Type XI collagenase overnight at 37°C for digestion. The digestion solution was filtered and centrifuged to obtain the chondrocyte pellets. The pellets were resuspended with growth medium (DMEM with 10% FBS and 1% penicillin-streptomycin mixture) and cultured at 37°C. For cell treatment, we used the human recombined cytokine IL-17A to induce CH. The human active CCL2 protein was used to upregulate cellular CCL2 expression. The CCL2 inhibitor (Bin) and CCR2 antagonist (CAS) were used to block CCL2 and CCR2 expression during IL-17A treatment.
IMMUNOFLUORESCENCE:
Protein expression of Type 10 collagen and CCL2 was analyzed with immunofluorescence (IF) methods using the following steps. Chondrocytes were seeded in a 24-well plate with a density of 7500 per well. After the drug treatments, the chondrocytes were washed using PBS and fixed with 4% paraformaldehyde for 10 min at room temperature. Then, the non-specific binding of the protein was blocked with 5% BSA for 1 h at room temperature. The chondrocytes then were incubated with the anti-Type 10 collagen and anti-CCL2 primary antibody overnight at 4°C. The next day, the chondrocytes were washed and incubated with the Alexa Fluor488 and 568 secondary antibodies for 1 h in the dark at room temperature. The nucleus was stained with 4′,6-diamidino-2-phenylindole (DAPI). The staining intensity was measured using Image-Pro Plus software to determine protein expression.
REAL-TIME PCR ANALYSIS:
We performed real-time PCR (RT-PCR) to analyze CCL2, CCR2, Type 2 collagen, Type 10 collagen, RUNX2, and IHH gene expression at the mRNA level. Briefly, total RNA was isolated from the chondrocytes after treatment using TRIzol reagent according to the manufacturer’s instructions. Then, the RNA was transcribed into complementary deoxyribose nucleic acid (cDNA) and applied to the RT-PCR analysis using Universal PCR Master Mix. The mixture contained 6 μL of RNase-free H2O, 12 μL of PCR Master Mix, and 2 μL cDNA. The reaction protocol was as follows: 95°C for 8 min, and 42 cycles at 95°C for 15 s, 60°C for 60 s, and 4°C for 5 min. Relative gene expression was normalized by the amount of GAPDH according using the 2−ΔΔCt method. The primers are listed in Table 1.
WESTERN BLOT:
The total protein in the chondrocytes was isolated using the RIPA buffer. An equal amount of protein from each group was separated with sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to the polyvinylidene fluoride membrane. The membranes were incubated with anti-Type 10 collagen, anti-CCL2, anti-CCR2, or GAPDH overnight at 4°C. After incubation with the secondary antibody, the membranes were exposed to enhanced chemiluminescence substrate and the band was measured using ImageJ software.
CELL AREA MEASUREMENT:
After treatment, the chondrocytes were washed 3 times with PBS and fixed with 4% paraformaldehyde for 15 min. Then, a white field image of the chondrocytes was examined under a microscope and the cell area was determined using ImageJ software.
FLOW CYTOMETRIC ANALYSIS:
Flow cytometry (FCM) was performed to identify the apoptotic chondrocytes in each group. The chondrocytes were harvested and resuspended in a 100-μL annexin-binding buffer. The Annexin V-FITC/PI kit was added to the cell suspension and incubated at 37°C for 15 min according to the manufacturer’s instructions. After incubation, emissions at 530 and 570 nm were used for FCM.
MTT ASSAY:
To assess the proliferation of chondrocytes after each treatment, we performed the MTT assay. Briefly, 104 chondrocytes were seeded in 96-well plates following the desired procedures. Viable cells were incubated with MTT assay kits according to the manufacturer’s instructions. The wavelength density of the spectrophotometric measurement was 570 nm.
STATISTICAL ANALYSIS:
Statistical analysis was performed using Statistical Product and Service Solutions (SPSS) software, version 22.0 (IBM, Armonk, New York, United States). The data are shown as means±standard deviations. Differences between 2 groups were analyzed by using a
Results
CCL2 AND CCR2 INCREASED IL-17A-INDUCED CHONDROCYTE HYPERTROPHY:
The proinflammatory cytokine IL-17 has been confirmed to enhance chondrocyte dedifferentiation [17]. We used a range of concentrations of IL-17A protein (10 ng/mL, 30 ng/mL, and 50 ng/mL) to incubate chondrocytes for 48 h and analyzed their chondrogenic or hypertrophic gene expression. The higher doses of IL-17A resulted in higher hypertrophic gene expression. After treatment with 30 ng/mL of IL-17A, Type 2 collagen expression was decreased and Type 10 collagen and Runx2 levels were increased compared to that in the control group. In contrast, results with the 50-ng/mL concentrated achieved more significance. The level of IHH mRNA was increased in a dose-dependent fashion, even with 10 ng/mL IL-17A treatment, compared to that in the control group (Figure 1A). Moreover, CCL2 and CCR2 expression were both upregulated after IL-17A stimulation in a dose-dependent manner compared with the control group (Figure 1B). Therefore, expression of CCL2 and its receptor, CCR2, was triggered when progression of CH began.
CCR2 DEFICIENCY DELAYED CCL2-INDUCED CH:
To determine the effects of CCL2 and CCR2 during CH, we supplied the CHs with active human CCL2 protein with and without CCR2 inhibition. The cells with no treatment were the control group. The efficiency of the inhibitors used in this study was analyzed at the protein level. After stimulation with Bin (50 μM) for 24 h, expression of CCL2 and CCR2 proteins was synchronously decreased compared with the controls. Treatment with CAS (5 nM) for 24 h significantly suppressed CCR2 protein levels (Figure 2A). Expression of CCL2 and CCR2 mRNA was measured to determine the optimal time of supplementation of the active human CCL2 protein. During the treatment with 10 ng/mL of CCL2 protein, cellular expression of CCL2 mRNA gradually increased for 48 h and then was stable from 48 to 72 h. In addition, we pretreated chondrocytes with 5 nM of CAS for 1 day and then switched to CCL2 incubation. The CAS pretreatment did not alter the CCL2 mRNA expression (Figure 2B). Unlike CCL2, expression of CCR2 mRNA in chondrocytes kept increasing during treatment with CCL2 protein. However, CCR2 expression in the CAS-pretreated chondrocytes was at a low level before rapidly increasing 24 h after CCL2 stimulation (Figure 2C). Therefore, we chose a 24-h CCL2 stimulation time to simultaneously obtain significant CCR2 inhibition. After 24 h of CCL2 protein treatment, Type 2 collagen expression decreased, which was reversed by CCR2 suppression. Expression of RUNX2 and IHH mRNA also was significantly increased due to the overexpression of CCL2, and the increment decreased when the chondrocytes were pretreated with CAS (Figure 2D). With the development of chondrocyte dedifferentiation, the proliferation decreased and the apoptosis usually increased. Given the results from MTT and FCM, CCL2 overexpression appeared to affect proliferation and promote apoptosis of the chondrocytes. Suppression of CCR2 by CAS, however, was integral to preventing the adverse effects caused by CCL2 (Figure 2E, 2F).
CCR2 DEFICIENCY INHIBITED CCL2-INDUCED TYPE X COLLAGEN EXPRESSION:
The cell area was measured to identify hypertrophic changes in chondrocytes. As shown in Figure 3A and 3B, CCL2 overexpression enlarged the cell area and resulted in a hypertrophic phenotype in CHs; pretreatment with a CCR2 inhibitor prevented this process. To assess the level of the hypertrophy markers Type 10 collagen and CCL2 in chondrocytes, we analyzed their protein expression with IF and western blot. As shown in Figure 3C and 3D, CCL2 protein expression was upregulated after supplementation with exogenic CCL2 protein, which was accompanied by increased Type 10 collagen protein production compared with controls. Moreover, suppression of CCR2 alleviated the CCL2-induced Type 10 collagen upregulation. However, CAS did not significantly affect CCL2 expression. The same trend was observed on the results of the western blots. CCL2 protein treatment increased the cellular expression of Type 10 collagen and CCL2 and pretreatment with CAS prevented upregulation of Type 10 collagen. CCL2 protein treatment also triggered upregulation of CCR2 protein expression, which was effectively suppressed by CAS (Figure 3E, 3F).
CCL2/CCR2 BLOCKADE INHIBITED IL-17A-INDUCED TYPE X COLLAGEN EXPRESSION:
Because overexpression of CCL2 accelerated CH, which could be interrupted by CCR2 deficiency, we wanted to investigate whether suppressing CCL2 or CCR2 could inhibit IL-17A-induced hypertrophy and if its efficiency could be amplified by combined CCL2 and CCR2 inhibition. Therefore, we pretreated chondrocytes with CAS (5 nM), Bin (50 μM), or a combination of CAS (5 nM) and Bin (50 μM) for 24 h. The chondrocytes then were cultured for 24 h with IL-17A (50 ng/mL) to induce hypertrophy, as previously described. The untreated chondrocytes served as controls. The cell morphology measurement results indicated that IL-17A significantly increased the cell area and that pretreatment with CAS, Bin, or both prevented the increase in cell area (Figure 4A, 4B). The IF results showed that CAS did not significantly affect CCL2 protein levels, but Bin pretreatment suppressed IL-17A-induced CCL2 upregulation. Furthermore, pretreatment with CAS, Bin, or the combination suppressed IL-17A-induced Type 10 collagen upregulation. The combination of CAS and Bin, however, was not more effective than treatment with either alone (Figure 4C, 4D). The inhibitory effects of CAS and Bin were verified using western blot, which suggested that suppression of CCL2 or CCR2 could alleviate IL-17A-induced Type 10 collagen upregulation, and that suppression of CCL2 resulted in a reduction in CCR2. Suppression of CCR2 did not affect the CCL2 level and combined treatment with CAS and Bin did not result in amplification (Figure 4E, 4F).
CCL2/CCR2 BLOCKING DELAYED IL-17A-INDUCED CHONDROCYTE HYPERTROPHY:
Collagen II expression also was protected after pretreatment with CAS, Bin, or CAS and Bin combined, compared to the IL-17A group. The mRNA of RUNX2 and IHH also was suppressed by inhibition of CCR2 or CCL2 (Figure 5A). Apart from this, suppression of CCR2 and CCL2 improved the proliferation and apoptosis triggered by IL-17A (Figure 5B, 5C). The efficiency of the destruction of IHH and apoptosis was more evident in the Bin-pretreated group than in the other 2 groups. However, the combination of CAS and Bin showed no advantage compared to treatment with either alone.
Discussion
In the present study, we investigated the role of the CCL2
First, we verified that CCL2 and CCR2 levels were increased when chondrocytes underwent hypertrophic differentiation caused by IL-17A. Moreover, exogenous supplementation of CCL2 also increased CCR2, which resulted in an enlarged cell area and hypertrophic markers in chondrocytes, which could be attenuated by blocking CCR2. Therefore, excessive CCL2 expression should be considered a risk factor for CH. In contrast to results from previous studies, we found that the effects of CCL2 on the morphology and hypertrophic differentiation of chondrocytes was related to upregulation of
Because CCL2 overexpression can induce CH, we explored the effects of inhibition of CCL2 on IL-17A-induced CH. CCL2 is a CC-type cell chemokine that induces the recruitment and activation of monocytes/macrophages by binding to CCR2 and it mediates most of the body’s inflammatory response processes. CCL2 and CCR2 have been proven to play an essential role in OA pathogenesis by promoting an inflammatory response and ECM degradation [24]. The molecular mechanism, however, has not been fully elucidated. For instance, Xu et al reported that CCL2 induced degradation of cartilage matrix and chondrocyte apoptosis by enhancing metalloproteinase [25]. Raghu et al stated that CCL2/CCR2 rather than the CCL5/CCR5 axis increased inflammation and tissue damage in OA [26]. In our study, we provided new insight related to the hypertrophy process in chondrocytes. Importantly, we found that pretreatment with an inhibitor of CCL2 or CCR2 resulted in significant resistance to IL-17A-induced hypertrophy, which means that suppressing the CCL2/CCR2 axis can prevent chondrocyte degeneration. This is in contrast to results from a previous study, which indicated that blocking CCL2 or CCR2 expression served as a treatment for inflammatory disease [27]. Our results also suggest that use of an CCL2/CCR2 axis inhibitor for prevention rather than treatment of OA is promising for alleviating pain and disability. In addition, we tested whether the combination of CAS and Bin could enhance the preventive effects, but no significant differences were observed. We speculate that: 1) CCL2 is the direct factor that induces CH and when it is inhibited, continued inhibition of CCR2 has no more influence; and 2) there might be a threshold value for inhibiting the CCL2/CCR2 axis that results in a delay in the hypertrophic process of chondrocytes.
The pathology of OA is complicated. Although has been confirmed to be a risk factor for it, our quantitative data elucidated the effects of the CCL2/CCR2 axis and its contribution to hypertrophy of chondrocytes. The significance of the present study is that it illuminated how pretreatment of chondrocytes with an inhibitor of CCL2 or CCR2 protects the chondrocytes from degeneration of CHs and revealed that the underlying mechanism is related to suppression of Type 10 collagen,
Conclusions
In conclusion, our work showed that the CCL2/CCR2 axis plays a crucial role in degeneration of chondrocytes. Blockade of the CCL2/CCR2 axis is meaningful to prevent the hypertrophic progress in chondrocytes, which is embodied in inhibition of Type 10 collagen,
Figures
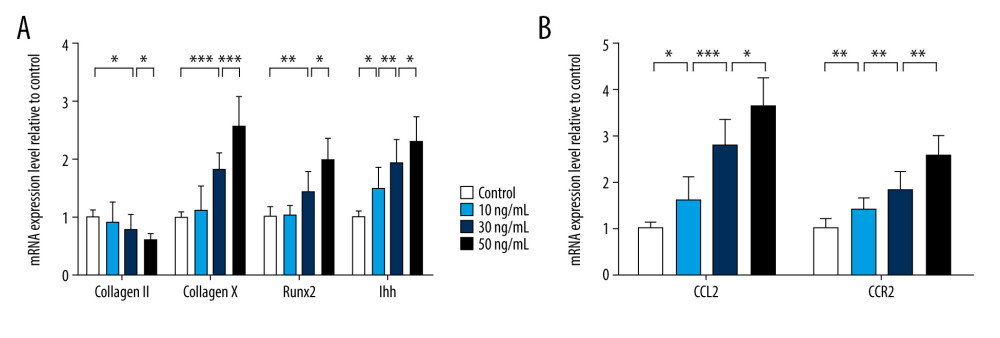 Figure 1. Chemokine (C-C motif) ligand 2 (CCL2) and CCR2 are increased in interleukin 17A-induced chondrocyte hypertrophy. Real-time polymerase chain reaction was used to assess mRNA expression of (A) Type 2 collagen, Type 10 collagen, RUNX2, and IHH and (B) CCL2 and CCR2. Data are shown as means±standard deviations vs the control group. * P<0.05, ** P<0.01, *** P<0.001.
Figure 1. Chemokine (C-C motif) ligand 2 (CCL2) and CCR2 are increased in interleukin 17A-induced chondrocyte hypertrophy. Real-time polymerase chain reaction was used to assess mRNA expression of (A) Type 2 collagen, Type 10 collagen, RUNX2, and IHH and (B) CCL2 and CCR2. Data are shown as means±standard deviations vs the control group. * P<0.05, ** P<0.01, *** P<0.001. 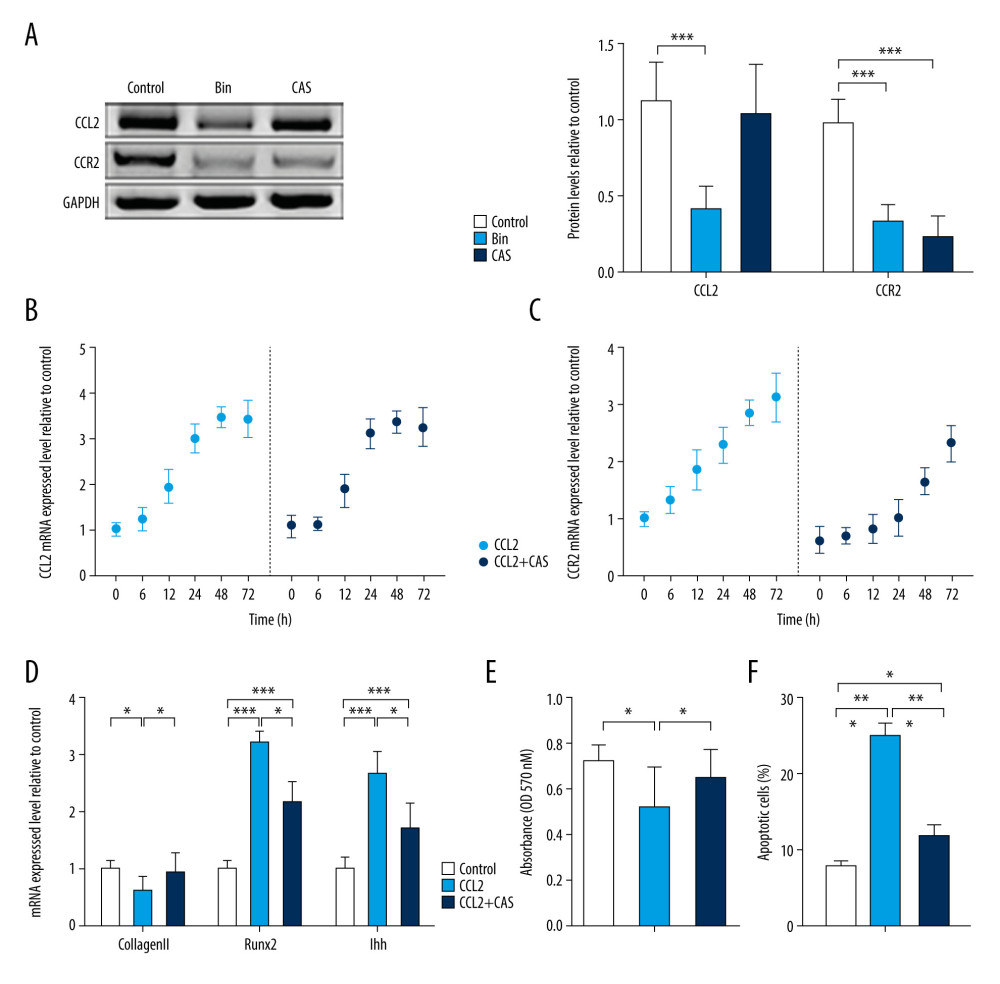 Figure 2. CCR2 deficiency delays chemokine (C-C motif) ligand 2 (CCL2)-induced chondrocyte hypertrophy (CH). (A) Protein expression of CCL2 and CCR2 was analyzed after treatment with bindarit or CAS and quantified. mRNA expression of (B) CCL2 and (C) CCR2 was analyzed after treatment with CCL2 protein with or without CAS pretreatment. (D) Expression of Type 2 collagen, RUNX2, and IHH mRNA in CH. (E) Cell proliferation was measured with an MTT assay. (F) Cell apoptosis was measured with flow cytometry. Data are shown as means±standard deviations vs the control group. * P<0.05, ** P<0.01, *** P<0.001.
Figure 2. CCR2 deficiency delays chemokine (C-C motif) ligand 2 (CCL2)-induced chondrocyte hypertrophy (CH). (A) Protein expression of CCL2 and CCR2 was analyzed after treatment with bindarit or CAS and quantified. mRNA expression of (B) CCL2 and (C) CCR2 was analyzed after treatment with CCL2 protein with or without CAS pretreatment. (D) Expression of Type 2 collagen, RUNX2, and IHH mRNA in CH. (E) Cell proliferation was measured with an MTT assay. (F) Cell apoptosis was measured with flow cytometry. Data are shown as means±standard deviations vs the control group. * P<0.05, ** P<0.01, *** P<0.001. 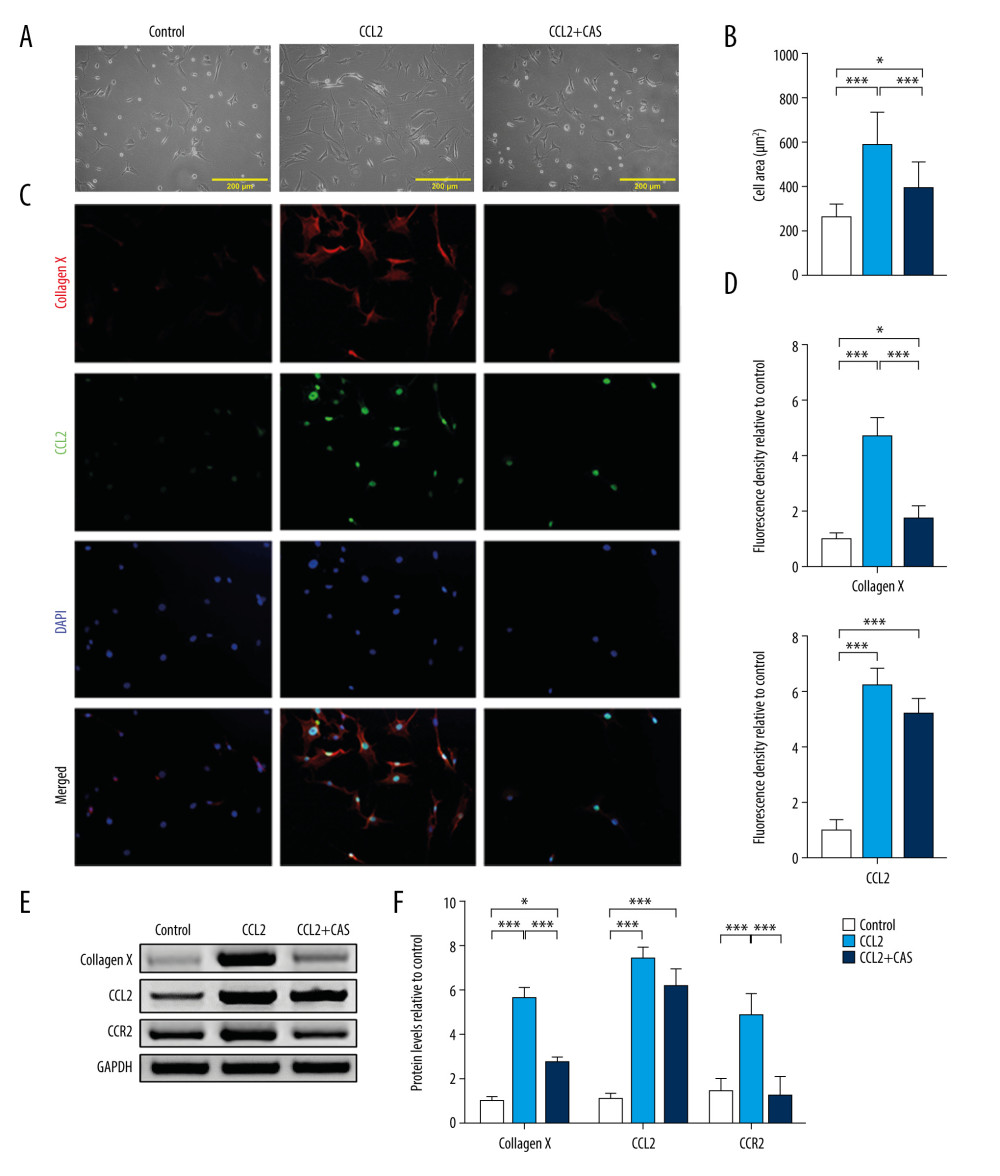 Figure 3. CCR2 deficiency inhibits chemokine (C-C motif) ligand 2 (CCL2)-induced Type 10 collagen expression. (A) Representative images of cell morphology and (B) cell area measurement. (C) Protein expression of Type 10 collagen and CCL2 was determined with immunofluorescence (IF) (magnification: 200×). Red indicates Type 10 collagen, green indicates CCL2, and blue indicates the cell nucleus. (D) Quantification analysis of IF intensity. (E) Protein expression of Type 10 collagen, CCL2, and CCR2 and (F) quantification analysis. Data are shown as means±standard deviations vs the control group. * P<0.05, *** P<0.001.
Figure 3. CCR2 deficiency inhibits chemokine (C-C motif) ligand 2 (CCL2)-induced Type 10 collagen expression. (A) Representative images of cell morphology and (B) cell area measurement. (C) Protein expression of Type 10 collagen and CCL2 was determined with immunofluorescence (IF) (magnification: 200×). Red indicates Type 10 collagen, green indicates CCL2, and blue indicates the cell nucleus. (D) Quantification analysis of IF intensity. (E) Protein expression of Type 10 collagen, CCL2, and CCR2 and (F) quantification analysis. Data are shown as means±standard deviations vs the control group. * P<0.05, *** P<0.001. 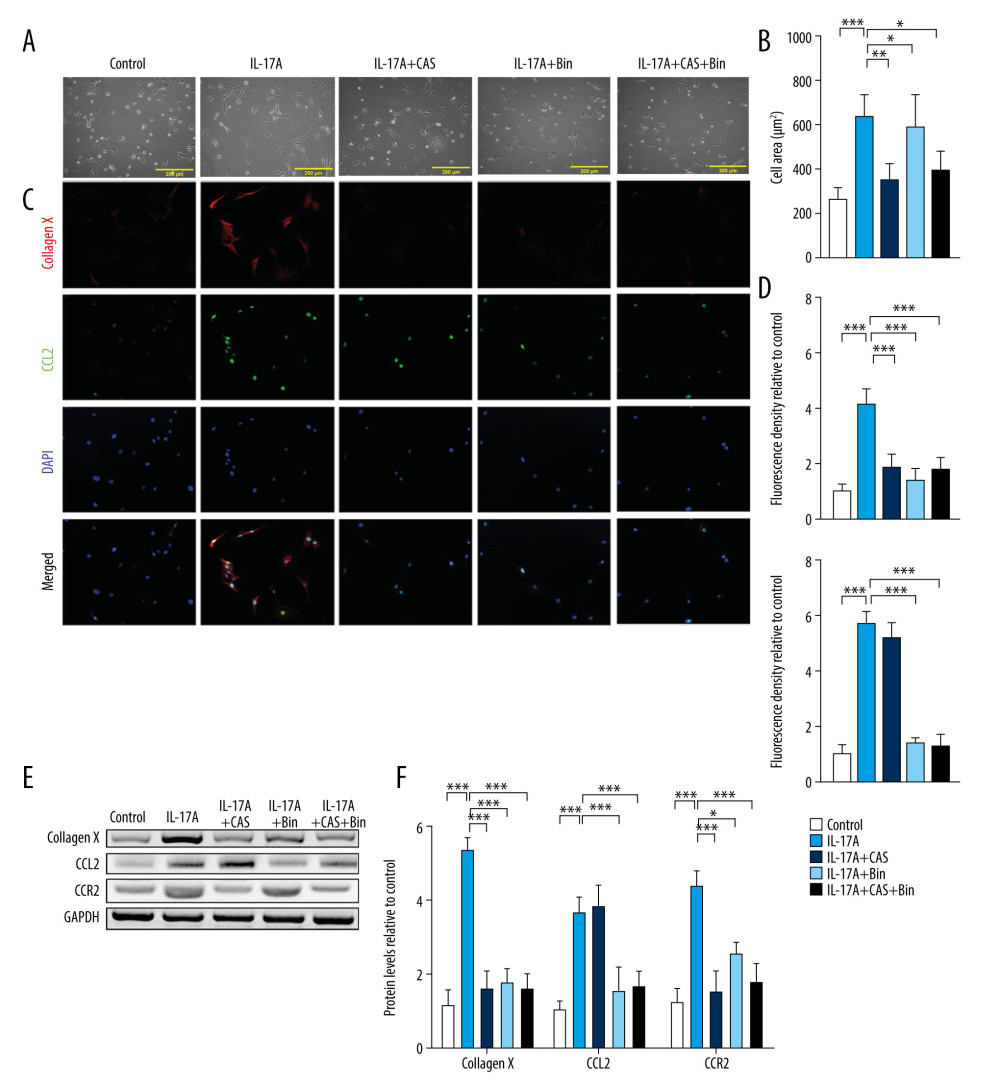 Figure 4. Blocking chemokine (C-C motif) ligand 2 (CCL2)/CCR2 inhibits interleukin-17A-induced Type 10 collagen expression. (A) Representative images of cell morphology and (B) cell area measurement. (C) Protein expression of Type 10 collagen and CCL2 was determined with immunofluorescence (IF) (magnification: 200×). Red indicates Type 10 collagen, green indicates CCL2, and blue indicates the cell nucleus. (D) Quantification analysis of IF intensity. (E) Protein expression of Type 10 collagen, CCL2, and CCR2 and (F) quantification analysis. Data are shown as means±standard deviations vs the control group. * P<0.05, *** P<0.001.
Figure 4. Blocking chemokine (C-C motif) ligand 2 (CCL2)/CCR2 inhibits interleukin-17A-induced Type 10 collagen expression. (A) Representative images of cell morphology and (B) cell area measurement. (C) Protein expression of Type 10 collagen and CCL2 was determined with immunofluorescence (IF) (magnification: 200×). Red indicates Type 10 collagen, green indicates CCL2, and blue indicates the cell nucleus. (D) Quantification analysis of IF intensity. (E) Protein expression of Type 10 collagen, CCL2, and CCR2 and (F) quantification analysis. Data are shown as means±standard deviations vs the control group. * P<0.05, *** P<0.001. 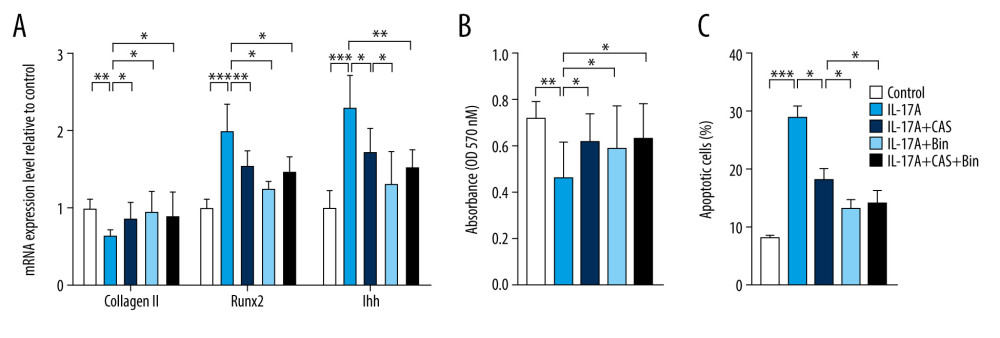 Figure 5. Blocking chemokine (C-C motif) ligand 2/CCR2 delays interleukin-17A-induced chondrocyte hypertrophy. (A) mRNA expression of Type 2 collagen, RUNX2, and IHH was assessed with real-time polymerase chain reaction. (B) Cell proliferation was measured with an MTT assay. (C) Cell apoptosis was measured with flow cytometry. Data are shown as means±standard deviations versus the control group. * P<0.05, ** P<0.01, *** P<0.001.
Figure 5. Blocking chemokine (C-C motif) ligand 2/CCR2 delays interleukin-17A-induced chondrocyte hypertrophy. (A) mRNA expression of Type 2 collagen, RUNX2, and IHH was assessed with real-time polymerase chain reaction. (B) Cell proliferation was measured with an MTT assay. (C) Cell apoptosis was measured with flow cytometry. Data are shown as means±standard deviations versus the control group. * P<0.05, ** P<0.01, *** P<0.001. 
References
1. Hunter DJ, Bierma-Zeinstra S, Osteoarthritis: Lancet, 2019; 393; 1745-59
2. Barranco C, Osteoarthritis: Activate autophagy to prevent cartilage degeneration?: Nat Rev Rheumatol, 2015; 11; 127
3. Vinatier C, Merceron C, Guicheux J, Osteoarthritis: From pathogenic mechanisms and recent clinical developments to novel prospective therapeutic options: Drug Discov Today, 2016; 21; 1932-37
4. Li G, Yin J, Gao J, Subchondral bone in osteoarthritis: Insight into risk factors and microstructural changes: Arthritis Res Ther, 2013; 15; 223
5. Scanzello CR, Umoh E, Pessler F, Local cytokine profiles in knee osteoarthritis: elevated synovial fluid interleukin-15 differentiates early from end-stage disease: Osteoarthritis Cartilage, 2009; 17; 1040-48
6. Wood S, Jayaraman V, Huelsmann EJ, Proinflammatory chemokine CCL2 (MCP-1) promotes healing in diabetic wounds by restoring the macrophage response: PLoS One, 2014; 9; e91574
7. Cerri C, Genovesi S, Allegra M, The chemokine CCL2 mediates the seizure-enhancing effects of systemic inflammation: J Neurosci, 2016; 36; 3777-88
8. Xu Z, Li J, Yang H, Association of CCL2 gene variants with osteoarthritis: Arch Med Res, 2019; 50; 86-90
9. Cecil DL, Johnson K, Rediske J, Inflammation-induced chondrocyte hypertrophy is driven by receptor for advanced glycation end products: J Immunol, 2005; 175; 8296-302
10. Borzi RM, Mazzetti I, Macor S, Flow cytometric analysis of intracellular chemokines in chondrocytes in vivo: Constitutive expression and enhancement in osteoarthritis and rheumatoid arthritis: Febs Lett, 1999; 455; 238-42
11. Longobardi L, Jordan JM, Shi XA, Associations between the chemokine biomarker CCL2 and knee osteoarthritis outcomes: The Johnston County Osteoarthritis Project: Osteoarthritis Cartilage, 2018; 26; 1257-61
12. Shin WS, Szuba A, Rockson SG, The role of chemokines in human cardiovascular pathology: Enhanced biological insights: Atherosclerosis, 2002; 160; 91-102
13. Baeck C, Wehr A, Karlmark KR, Pharmacological inhibition of the chemokine CCL2 (MCP-1) diminishes liver macrophage infiltration and steatohepatitis in chronic hepatic injury: Gut, 2012; 61; 416-26
14. Madhavan S, Anghelina M, Rath-Deschner B, Biomechanical signals exert sustained attenuation of proinflammatory gene induction in articular chondrocytes: Osteoarthritis Cartilage, 2006; 14; 1023-32
15. Han R, Gu S, Zhang Y, Estrogen promotes progression of hormone-dependent breast cancer through CCL2-CCR2 axis by upregulation of Twist via PI3K/AKT/NF-kappaB signaling: Sci Rep, 2018; 8; 9575
16. Bay-Jensen AC, Thudium CS, Mobasheri A, Development and use of biochemical markers in osteoarthritis: Current update: Curr Opin Rheumatol, 2018; 30; 121-28
17. Honorati MC, Cattini L, Facchini A, IL-17, IL-1beta and TNF-alpha stimulate VEGF production by dedifferentiated chondrocytes: Osteoarthritis Cartilage, 2004; 12; 683-91
18. Papaioannou G, Mirzamohammadi F, Lisse TS, MicroRNA-140 provides robustness to the regulation of hypertrophic chondrocyte differentiation by the PTHrP-HDAC4 pathway: J Bone Miner Res, 2015; 30; 1044-52
19. Bhattacharjee M, Coburn J, Centola M, Tissue engineering strategies to study cartilage development, degeneration and regeneration: Adv Drug Deliv Rev, 2015; 84; 107-22
20. Madhavan S, Anghelina M, Rath-Deschner B, Biomechanical signals exert sustained attenuation of proinflammatory gene induction in articular chondrocytes: Osteoarthritis Cartilage, 2006; 14; 1023-32
21. Amano K, Densmore M, Nishimura R, Indian hedgehog signaling regulates transcription and expression of collagen type X via Runx2/Smads interactions: J Biol Chem, 2014; 289; 24898-910
22. Catheline SE, Hoak D, Chang M, Chondrocyte-specific Runx2 overexpression accelerates post-traumatic osteoarthritis progression in adult mice: J Bone Miner Res, 2019; 34; 1676-89
23. Dong YF, Soung DY, Schwarz EM, Wnt induction of chondrocyte hypertrophy through the Runx2 transcription factor: J Cell Physiol, 2006; 208; 77-86
24. Melgarejo E, Medina MA, Sanchez-Jimenez F, Monocyte chemoattractant protein-1: A key mediator in inflammatory processes: Int J Biochem Cell Biol, 2009; 41; 998-1001
25. Xu YK, Ke Y, Wang B, The role of MCP-1-CCR2 ligand-receptor axis in chondrocyte degradation and disease progress in knee osteoarthritis: Biol Res, 2015; 48; 64
26. Raghu H, Lepus CM, Wang Q, CCL2/CCR2, but not CCL5/CCR5, mediates monocyte recruitment, inflammation and cartilage destruction in osteoarthritis: Ann Rheum Dis, 2017; 76; 914-22
27. Jiang S, Wang Q, Wang Y, Blockade of CCL2/CCR2 signaling pathway prevents inflammatory monocyte recruitment and attenuates OVA-Induced allergic asthma in mice: Immunol Lett, 2019; 214; 30-36
Figures
Figure 1. Chemokine (C-C motif) ligand 2 (CCL2) and CCR2 are increased in interleukin 17A-induced chondrocyte hypertrophy. Real-time polymerase chain reaction was used to assess mRNA expression of (A) Type 2 collagen, Type 10 collagen, RUNX2, and IHH and (B) CCL2 and CCR2. Data are shown as means±standard deviations vs the control group. * P<0.05, ** P<0.01, *** P<0.001.Figure 2. CCR2 deficiency delays chemokine (C-C motif) ligand 2 (CCL2)-induced chondrocyte hypertrophy (CH). (A) Protein expression of CCL2 and CCR2 was analyzed after treatment with bindarit or CAS and quantified. mRNA expression of (B) CCL2 and (C) CCR2 was analyzed after treatment with CCL2 protein with or without CAS pretreatment. (D) Expression of Type 2 collagen, RUNX2, and IHH mRNA in CH. (E) Cell proliferation was measured with an MTT assay. (F) Cell apoptosis was measured with flow cytometry. Data are shown as means±standard deviations vs the control group. * P<0.05, ** P<0.01, *** P<0.001.Figure 3. CCR2 deficiency inhibits chemokine (C-C motif) ligand 2 (CCL2)-induced Type 10 collagen expression. (A) Representative images of cell morphology and (B) cell area measurement. (C) Protein expression of Type 10 collagen and CCL2 was determined with immunofluorescence (IF) (magnification: 200×). Red indicates Type 10 collagen, green indicates CCL2, and blue indicates the cell nucleus. (D) Quantification analysis of IF intensity. (E) Protein expression of Type 10 collagen, CCL2, and CCR2 and (F) quantification analysis. Data are shown as means±standard deviations vs the control group. * P<0.05, *** P<0.001.Figure 4. Blocking chemokine (C-C motif) ligand 2 (CCL2)/CCR2 inhibits interleukin-17A-induced Type 10 collagen expression. (A) Representative images of cell morphology and (B) cell area measurement. (C) Protein expression of Type 10 collagen and CCL2 was determined with immunofluorescence (IF) (magnification: 200×). Red indicates Type 10 collagen, green indicates CCL2, and blue indicates the cell nucleus. (D) Quantification analysis of IF intensity. (E) Protein expression of Type 10 collagen, CCL2, and CCR2 and (F) quantification analysis. Data are shown as means±standard deviations vs the control group. * P<0.05, *** P<0.001.Figure 5. Blocking chemokine (C-C motif) ligand 2/CCR2 delays interleukin-17A-induced chondrocyte hypertrophy. (A) mRNA expression of Type 2 collagen, RUNX2, and IHH was assessed with real-time polymerase chain reaction. (B) Cell proliferation was measured with an MTT assay. (C) Cell apoptosis was measured with flow cytometry. Data are shown as means±standard deviations versus the control group. * P<0.05, ** P<0.01, *** P<0.001. In Press
Clinical Research
Effects of Single-Bout Endurance Exercise Intensity on Peripheral Neurotrophic Factors in Patients With Isc...Med Sci Monit In Press; DOI: 10.12659/MSM.952089
Review article
Anisodus tanguticus in Cancer Research: A Review of Traditional Use, Phytochemistry, Extraction Methods, an...Med Sci Monit In Press; DOI: 10.12659/MSM.952999
Clinical Research
Nasal Mucociliary Clearance and Its Relationship With Disease Severity in Patients With Multiple SclerosisMed Sci Monit In Press; DOI: 10.12659/MSM.952850
Clinical Research
Modified Thoracoabdominal Nerves Block Through the Perichondrial Approach vs Subcostal Transversus Abdomini...Med Sci Monit In Press; DOI: 10.12659/MSM.953976
Most Viewed Current Articles
17 Jan 2024 : Review article 14,176,570
Vaccination Guidelines for Pregnant Women: Addressing COVID-19 and the Omicron VariantDOI :10.12659/MSM.942799
Med Sci Monit 2024; 30:e942799
13 Nov 2021 : Clinical Research 3,762,188
Acceptance of COVID-19 Vaccination and Its Associated Factors Among Cancer Patients Attending the Oncology ...DOI :10.12659/MSM.932788
Med Sci Monit 2021; 27:e932788
14 Dec 2022 : Clinical Research 2,466,310
Prevalence and Variability of Allergen-Specific Immunoglobulin E in Patients with Elevated Tryptase LevelsDOI :10.12659/MSM.937990
Med Sci Monit 2022; 28:e937990
16 May 2023 : Clinical Research 708,927
Electrophysiological Testing for an Auditory Processing Disorder and Reading Performance in 54 School Stude...DOI :10.12659/MSM.940387
Med Sci Monit 2023; 29:e940387